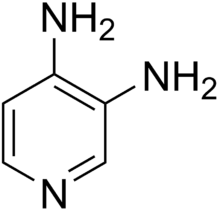
Amifampridine
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Firdapse |
| AHFS/Drugs.com | UK Drug Information |
| License data | |
| Pregnancy category |
|
| Routes of administration | Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 93–100%[1] |
| Metabolism | Acetylation to 3-N-acetylamifampridine |
| Elimination half-life |
2.5 hrs (amifampridine) 4 hrs (3-N-acetylamifampridine) |
| Excretion | Renal (19% unmetabolized, 74–81% 3-N-acetylamifampridine |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard |
100.000.201 |
| Chemical and physical data | |
| Formula | C5H7N3 |
| Molar mass | 109.13 g·mol−1 |
| 3D model (JSmol) | |
| Melting point | 218 to 220 °C (424 to 428 °F) decomposes |
| Solubility in water | 24 mg/mL (20 °C) |
| |
| |
| | |
Amifampridine (pyridine-3,4-diamine, 3,4-diaminopyridine, 3,4-DAP) is used as a drug, predominantly in the treatment of a number of rare muscle diseases. The free base form of the drug has been used to treat congenital myasthenic syndromes and Lambert–Eaton myasthenic syndrome (LEMS) through compassionate use programs since the 1990s and was recommended as a first line treatment for LEMS in 2006, using ad hoc forms of the drug, since there was no marketed form.
Around 2000 doctors at Assistance Publique – Hôpitaux de Paris created a phosphate salt form, which was developed through a series of companies ending with BioMarin Pharmaceutical which obtained European approval in 2009 under the trade name Firdapse, and which licensed the US rights to Catalyst Pharmaceuticals in 2012. As of January 2017, Catalyst and another US company, Jacobus Pharmaceuticals, which had been manufacturing the free base form and giving it away for free since the 1990s, were racing to obtain FDA approval for their versions first; the company that obtained the approval would have seven years of marketing exclusivity.
Amifampridine phosphate has orphan drug status in the EU for Lambert–Eaton myasthenic syndrome and Catalyst holds both an orphan designation and a breakthrough therapy designation in the US.
Medical uses
Amifampridine is used to treat many of the congenital myasthenic syndromes, particularly those with defects in choline acetyltransferase, downstream kinase 7, and those where any kind of defect causes "fast channel" behaviour of the acetylcholine receptor.[2][3] It is also used to treat psoriasis. It is also used to treat symptoms of Lambert–Eaton myasthenic syndrome.[1][4]
Contraindications
Because it affects voltage-gated ion channels, it is contraindicated in people with long QT syndrome and in people taking a drug that might prolong QT like sultopride, disopyramide, cisapride, domperidone, rifampicin or ketoconazol. It is also contraindicated in people with epilepsy or badly controlled asthma.[1]
Adverse effects
The dose-limiting side effects include tingling or numbness, difficulty sleeping, fatigue, and loss of muscle strength.[5]
Interactions via the liver's cytochrome P450 enzyme system are considered unlikely.[1]
Pharmacology
Mechanism of action
In Lambert–Eaton myasthenic syndrome, acetylcholine release is inhibited as antibodies involved in the host response against certain cancers cross-react with Ca2+ channels on the prejunctional membrane. Amifampridine works by blocking potassium channel efflux in nerve terminals so that action potential duration is increased.[6] Ca2+ channels can then be open for a longer time and allow greater acetylcholine release to stimulate muscle at the end plate.[5]
Pharmacokinetics
Amifampridine is quickly and almost completely (93–100%) absorbed from the gut. In a study with 91 healthy subjects, maximum amifampridine concentrations in blood plasma were reached after 0.6 (±0.25) hours when taken without food, or after 1.3 (±0.9) hours after a fatty meal, meaning that the speed of absorption varies widely. Biological half-life (2.5±0.7 hrs) and the area under the curve (AUC = 117±77 ng∙h/ml) also vary widely between subjects, but are nearly independent of food intake.[1]
The substance is deactivated by acetylation via N-acetyltransferases to the single metabolite 3-N-acetylamifampridine. Activity of these enzymes (primarily N-acetyltransferase 2) in different individuals seems to be primarily responsible for the mentioned differences in half-life and AUC: the latter is increased up to 9-fold in slow metabolizers as compared to fast metabolizers.[1]
Amifampridine is eliminated via the kidneys and urine to 74–81% as N-acetylamifampridine and to 19% in unchanged form.[1]
Chemistry
3,4-Diaminopyridine is a pale yellow to pale brown crystalline powder that melts at about 218–220 °C (424–428 °F) under decomposition. It is readily soluble in methanol, ethanol and hot water, but only slightly in diethyl ether.[7][8] Solubility in water at 20 °C (68 °F) is 25 g/L.
The drug formulation amifampridine phosphate contains the phosphate salt, more specifically 4-aminopyridine-3-ylammonium dihydrogen phosphate.[8] This salt forms prismatic, monoclinic crystals (space group C2/c)[9] and is readily soluble in water.[10] The phosphate salt is stable, and does not require refrigeration.[11]
History
The development of amifampridine and its phosphate has brought attention to orphan drug policies that grant market exclusivity as an incentive for companies to develop therapies for conditions that affect small numbers of people.[12][13][14]
Amifampridine, also called 3,4-DAP, was discovered in Scotland in the 1970s, and doctors in Sweden first showed its use in LEMS in the 1980s.[15]
In the 1990s, doctors in the US, on behalf of Muscular Dystrophy Association, approached a small family-owned manufacturer of active pharmaceutical ingredients in New Jersey, Jacobus Pharmaceuticals, about manufacturing amifampridine so they could test it in clinical trials. Jacobus did so, and when the treatment turned out to be effective, Jacobus and the doctors were faced with a choice — invest in clinical trials to get FDA approval or give the drug away for free under a compassionate use program. Jacobus elected to give the drug away, and did so for about twenty years.[16][17]
Doctors at the Assistance Publique – Hôpitaux de Paris had created a phosphate salt of 3,4-DAP (3,4-DAPP), and obtained an orphan designation for it in Europe in 2002.[18] The hospital licensed the intellectual property on the phosphate form to the French biopharma company OPI, which was acquired by EUSA Pharma in 2007,[19] and the orphan application was transferred to EUSA in 2008.[18] In 2008 EUSA submitted an application for approval to market the phosphate form to the European Medicines Agency under the brand name, Zenas.[20] EUSA, through a vehicle called Huxley Pharmaceuticals, sold the rights to 3,4-DAPP to Biomarin in 2009,[21] the same year that 3,4-DAPP was approved in Europe under the new name Firdapse.[18] BioMarin launched the drug in Europe in April 2010 and through the first nine months of 2012 year had sales of $10.8 million.[22]
The licensing of Firdapse in 2010 in Europe led to a sharp increase in price for the drug. In some cases, this has led to hospitals using an unlicensed form rather than the licensed agent, as the price difference proved prohibitive. BioMarin has been criticized for licensing the drug on the basis of previously conducted research, and yet charging exorbitantly for it.[23] A group of UK neurologists and pediatricians petitioned to prime minister David Cameron in an open letter to review the situation.[24] The company responded that it submitted the licensing request at the suggestion of the French government, and points out that the increased cost of a licensed drug also means that it is monitored by regulatory authorities (e.g. for uncommon side effects), a process that was previously not present in Europe.[25] A 2011 Cochrane review compared the cost of the 3,4-DAP and 3,4-DAPP in the UK and found an average price for 3,4-DAP base of £1/tablet and an average price for 3,4-DAP phosphate of £20/tablet; and the authors estimated a yearly cost per person of £730 for the base versus £29,448 for the phosphate formulation.[4][11]
Meanwhile, in Europe, a task force of neurologists had recommended 3,4-DAP as the firstline treatment for LEMS symptoms in 2006, even though there was no approved form for marketing; it was being supplied ad hoc.[20]:5[26] In 2007 the drug's international nonproprietary name was published by the WHO.[27]
In the US, an orphan drug designation for 3,4-DAPP had been granted in 2009[28] and clinical trials were well under way by 2012.[22]
In the face of the 7 year exclusivity that an orphan approval would give to Biomarin, and of the increase in price that would accompany it, Jacobus began racing to conduct formal clinical trials in order to get approval for the free base form before BioMarin; its first Phase II trial was opened in January 2012.[29]
In October 2012, while BioMarin had a Phase III trial ongoing in the US, it licensed the US rights to 3,4-DAPP, including the orphan designation and the ongoing trial, to Catalyst Pharmaceuticals.[22] Catalyst anticipated that it could earn $300 to $900 million per year in sales for treatment of people with LEMS and other indications, and analysts anticipated the drug would be priced at around. $100,000 in the US.[15] Catalyst went on to obtain a breakthrough therapy designation for 3,4-DAPP in LEMS in 2013,[30] an orphan designation for congenital myasthenic syndromes in 2015[31] and an orphan designation for myasthenia gravis in 2016.[32]
In August 2013, analysts anticipated that FDA approval would be granted to Catalyst in LEMS by 2015.[33]
In October 2014, Catalyst began making available under an expanded access program.[34]
In March 2015 Catalyst obtained an orphan designation for the use of 3,4-DAPP to treat of congenital myasthenic syndromes, causing its stock to rise a bit.[35][36] In April 2015, Jacobus presented clinical trial results with 3,4-DAP at a scientific meeting, causing Catalyst shares to fall by 42%.[17]
In December 2015 a group of 106 neuromuscular doctors who had worked with both Jacobus and BioMarin/Catalyst published an editorial in the journal, Muscle & Nerve, expressing concern about the potential for the price of the drug to be dramatically increased should Catalyst obtain FDA approval, and stating that 3,4-DAPP represented no real innovation and didn't deserve exclusivity under the Orphan Drug Act, which was meant to spur innovation to meet unmet needs.[15][37] Other commentators noted, however, that Jacobus was not able to meet the needs of all people who could benefit from the drug via its free program, and that its manufacturing practices had quality control problems, with 483 FDA citations in 2011 and 2012.[16] Similarly, at the urging of Assistance Publique-Hôpitaux de Paris, the French drug authority had reviewed the use of the free base in light of the manufactured phosphate form in 2006, and had urged doctors in France not to use the free base form due to quality issues.[38]
In December 2015 Catalyst submitted its new drug application to the FDA,[39] and in February 2016 the FDA refused to accept it, on the basis that it wasn't complete and in April 2016 the FDA told Catalyst it would have to gather further data.[40][12] Catalyst cut 30% of its workforce to save money to conduct the trials.[41] In March 2018 the company re-submitted its NDA.[42]
Research
Amifampridine has also been proposed for the treatment of multiple sclerosis (MS). A 2002 Cochrane systematic review found that there was no unbiased data to support its use for treating MS.[43] There was no change as of 2012.[44]
See also
References
- 1 2 3 4 5 6 7 "Firdapse Summary of Product Characteristics" (PDF). EMA. February 11, 2010. See EMA Index page, product tab
- ↑ Argov Z (Oct 2009). "Management of myasthenic conditions: nonimmune issues". Current Opinion in Neurology. 22 (5): 493–7. doi:10.1097/WCO.0b013e32832f15fa. PMID 19593127.
- ↑ Abicht, Angela; Müller, Juliane; Lochmüller, Hanns (July 14, 2016). "Congenital Myasthenic Syndromes". GeneReviews. University of Washington, Seattle.
- 1 2 Keogh M, Sedehizadeh S, Maddison P (2011). "Treatment for Lambert-Eaton myasthenic syndrome". The Cochrane Database of Systematic Reviews (2): CD003279. doi:10.1002/14651858.CD003279.pub3. PMID 21328260.
- 1 2 Tarr, TB; Wipf, P; Meriney, SD (August 2015). "Synaptic Pathophysiology and Treatment of Lambert-Eaton Myasthenic Syndrome". Molecular neurobiology. 52 (1): 456–63. doi:10.1007/s12035-014-8887-2. PMC 4362862. PMID 25195700.
- ↑ Kirsch GE, Narahashi T (June 1978). "3,4-diaminopyridine. A potent new potassium channel blocker". Biophys J. 22 (3): 507–12. doi:10.1016/s0006-3495(78)85503-9. PMC 1473482. PMID 667299.
- ↑ "Diaminopyridine (3,4-)" (PDF). FDA. Retrieved 28 November 2015. . Index page: FDA Docket 98N-0812: Bulk Drug Substances to be Used in Pharmacy Compounding
- 1 2 Dinnendahl, V; Fricke, U, eds. (2015). Arzneistoff-Profile (in German). 1 (28th ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 978-3-7741-9846-3.
- ↑ "Crystal Structure and Solid-State Properties of 3,4-Diaminopyridine Dihydrogen Phosphate and Their Comparison with Other Diaminopyridine Salts". Cryst Growth Des. 13 (2): 708–715. 2013. doi:10.1021/cg3014249.
- ↑ A. Klement (9 November 2015). "Firdapse". Österreichische Apothekerzeitung (in German) (23/2015): 10f.
- 1 2 "Evidence Review: Amifampridine phosphate for the treatment of Lambert–Easton Myasthenic Syndrome" (PDF). NHS England. December 2015.
- 1 2 Tavernise, Sabrina (February 17, 2016). "F.D.A. Deals Setback to Catalyst in Race for Drug Approval". New York Times.
- ↑ Drummond, M; Towse, A (May 2014). "Orphan drugs policies: a suitable case for treatment". The European journal of health economics : HEPAC : health economics in prevention and care. 15 (4): 335–40. doi:10.1007/s10198-014-0560-1. PMID 24435513.
- ↑ Lowe, Derek (21 October 2013). "Catalyst Pharmaceuticals And Their Business Plan". In the Pipeline.
- 1 2 3 Deak, Dalia (February 22, 2016). "Jacobus and Catalyst Continue to Race for Approval of LEMS Drug". Bill of Health.
- 1 2 Silverman, Ed (5 April 2016). "A family-run drug maker tries to stay afloat in the Shkreli era". STAT News.
- 1 2 "Jacobus Pharmaceuticals". Drug R&D Insight. April 25, 2015.
- 1 2 3 "Public summary of opinion on orphan designation" (PDF). EMA. 14 June 2010.
- ↑ Chapelle, François-Xavier (November 4, 2008). "OPi ou comment construire une biopharma en moins de dix ans - Private Equity Magazine". Private Equity Magazine (in French).
- 1 2 "Assessment report: Zenas" (PDF). EMA CHMP committee. 2009.
- ↑ "Huxley Acquisition Lands Biomarin New LEMS Treatment". Pharmaceutical Technology. October 28, 2009.
- 1 2 3 Leuty, Ron (October 31, 2012). "BioMarin licenses North American rights to rare disease drug, invests $5M in Florida company". San Francisco Business Journal.
- ↑ Goldberg, Adrian (21 November 2010). "Drug firms accused of exploiting loophole for profit". BBC News.
- ↑ Nicholl DJ, Hilton-Jones D, Palace J, Richmond S, Finlayson S, Winer J, Weir A, Maddison P, Fletcher N, Sussman J, Silver N, Nixon J, Kullmann D, Embleton N, Beeson D, Farrugia ME, Hill M, McDermott C, Llewelyn G, Leonard J, Morris M (2010). "Open letter to prime minister David Cameron and health secretary Andrew Lansley". BMJ. 341: c6466. doi:10.1136/bmj.c6466. PMID 21081599.
- ↑ Hawkes N, Cohen D (2010). "What makes an orphan drug?". BMJ. 341: c6459. doi:10.1136/bmj.c6459. PMID 21081607.
- ↑ Vedeler, CA; Antoine, JC; Giometto, B; Graus, F; Grisold, W; Hart, IK; Honnorat, J; Sillevis Smitt, PA; Verschuuren, JJ; Voltz, R; Paraneoplastic Neurological Syndrome Euronetwork. (July 2006). "Management of paraneoplastic neurological syndromes: report of an EFNS Task Force". European Journal of Neurology. 13 (7): 682–90. doi:10.1111/j.1468-1331.2006.01266.x. PMID 16834698.
- ↑ "International Nonproprietary Names for Pharmaceutical Substances (INN) Recommended INN: List 58" (PDF). WHO Drug Information. 21 (3). 2007.
- ↑ "Orphan Drug Designations for amifampridine phosphate for LAMS". www.accessdata.fda.gov. FDA. Retrieved 14 January 2017.
- ↑ Wahl, Margaret (January 25, 2012). "Jacobus Begins Invitation-Only Trial of 3,4-DAP in LEMS". Muscular Dystrophy Association Quest Magazine Online.
- ↑ Baker DE (Nov 2013). "Breakthrough Drug Approval Process and Postmarketing ADR Reporting". Hospital Pharmacy. 48 (10): 796–8. doi:10.1310/hpj4810-796. PMC 3859287. PMID 24421428.
- ↑ "Orphan Drug Designations: amifampridine phosphate for congenital myasthenic syndromes". FDA. Retrieved 14 January 2017.
- ↑ "Orphan Drug Designations: amifampridine phosphate for myasthenia gravis". www.accessdata.fda.gov. FDA. Retrieved 14 January 2017.
- ↑ Bandell B (August 28, 2013). "Shares of Catalyst Pharmaceuticals soar 42% on FDA news". South Florida Business Journal.
- ↑ Radke, James (October 29, 2014). "Catalyst Using the Expanded Access Program to Conduct Phase IV Study with LEMS Patients". Rare Disease Report.
- ↑ "Orrphan designation congenital myasthenic syndromes". FDA. Archived from the original on July 26, 2015.
- ↑ Ampel, Celia (5 March 2015). "Catalyst Pharma stock rises 5% after firm hits FDA milestone". South Florida Business Journal.
- ↑ Burns, TM, et al.I (February 2016). "Editorial by concerned physicians: Unintended effect of the orphan drug act on the potential cost of 3,4-diaminopyridine". Muscle & nerve. 53 (2): 165–8. doi:10.1002/mus.25009. PMID 26662952.
- ↑ "Point d'information sur l'utilisation de la 3.4 diaminopyridine". ANSM : Agence nationale de sécurité du médicament et des produits de santé. December 18, 2006.
- ↑ Tavernise, Sabrina (December 22, 2015). "Patients Fear Spike in Price of Old Drugs". New York Times.
- ↑ Adams, Ben (April 26, 2016). "Catalyst Pharmaceuticals hit by FDA extra studies request for Firdapse". FierceBiotech.
- ↑ Adams, Ben (May 17, 2016). "Catalyst to ax 30% of workforce in wake of FDA trial demands". FierceBiotech.
- ↑ Lima, Debora (March 29, 2018). "Catalyst Pharmaceuticals files new drug application with FDA". South Florida Business Journa.
- ↑ Solari, A.; Uitdehaag, B.; Giuliani, G.; Pucci, E.; Taus, C. (2002). "Aminopyridines for symptomatic treatment in multiple sclerosis". The Cochrane Database of Systematic Reviews (4): CD001330. doi:10.1002/14651858.CD001330. ISSN 1469-493X. PMID 12804404.
- ↑ Sedehizadeh, S; Keogh, M; Maddison, P (2012). "The use of aminopyridines in neurological disorders". Clinical neuropharmacology. 35 (4): 191–200. doi:10.1097/WNF.0b013e31825a68c5. PMID 22805230.