Romano–Ward syndrome
| Romano–Ward syndrome | |
|---|---|
 | |
| Schematic representation of normal ECG trace (sinus rhythm), with waves, segments, and intervals labeled. | |
| Causes | Mutations in the KCNQ1, KCNH2, and SCN5A genes [1] |
| Diagnostic method | EKG, Exercise test[2] |
| Treatment | Beta-adrenergic blockade [3] |
Romano–Ward syndrome is the major variant of long QT syndrome,[4][1] a heart condition that causes the cardiac muscle to take longer than usual to recharge between beats; if untreated, the irregular heartbeats can lead to fainting, seizures, or sudden death.[5]
Signs and symptoms
Romano–Ward syndrome presents the following in an affected individual:[6]
- Ventricular fibrillation
- Syncope
- Torsade de pointes
- Abnormality of ear
Genetics
Romano–Ward syndrome is inherited in an autosomal dominant pattern. It is the most common form of inherited long QT syndrome, affecting an estimated 1 in 7,000 people worldwide. Romano-Ward syndrome is one of six variations of long QT syndrome.[1][6]
Mechanism
The mechanism of Romano–Ward syndrome sees that mutations in the ANK2, KCNE1, KCNE2, KCNH2, KCNQ1, and SCN5A genes can cause Romano–Ward syndrome. The proteins made by most of these genes form channels that transport positively-charged ions, such as K+ and Na+. In cardiac muscle, these ion channels play critical roles in maintaining the heart's normal rhythm. Mutations in any of these genes, for example ANK2, KCNE1, KCNE2, alter the structure or function of channels, which changes the flow of ions between cells, a disruption in ion transport alters the way the heart beats, leading to abnormal heart rhythm characteristic of the syndrome.[3][7][8][9]
The protein made by the ANK2 gene ensures that other proteins, particularly ion channels, are inserted into the cell membrane appropriately. A mutation in the ANK2 gene likely alters the flow of ions between cells in the heart, which disrupts the heart's normal rhythm and results in the features of Romano–Ward syndrome.
Diagnosis
In terms of the diagnosis of Romano–Ward syndrome the following is done to ascertain the condition (the Schwartz Score helps in so doing):[2]
Treatment
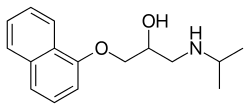
Treatment for Romano–Ward syndrome can deal with the imbalance between the right and left sides of the sympathetic nervous system which may play a role in the cause of this syndrome. The imbalance can be temporarily abolished with a left stellate ganglion block, which shorten the QT interval. If this is successful, surgical ganglionectomy can be performed as a permanent treatment.[10]Ventricular dysrhythmia may be managed by beta-adrenergic blockade (propranolol)[11][3]
See also
References
- 1 2 3 Reference, Genetics Home. "Romano-Ward syndrome". Genetics Home Reference. Retrieved 2017-04-01.
- 1 2 Mizusawa, Yuka; Horie, Minoru; Wilde, Arthur AM (2014-01-01). "Genetic and Clinical Advances in Congenital Long QT Syndrome". Circulation Journal. 78 (12): 2827–2833. doi:10.1253/circj.CJ-14-0905.
- 1 2 3 RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Romano Ward syndrome". www.orpha.net. Retrieved 2017-04-01.
- ↑ "Long QT syndrome 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-04-17.
- ↑ Alders, Mariëlle; Christiaans, Imke (1993-01-01). Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Ledbetter, Nikki; Mefford, Heather C., eds. GeneReviews(®). Seattle (WA): University of Washington, Seattle. PMID 20301308. update 2015
- 1 2 "Long QT syndrome 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2017-04-01.
- ↑ "ANK2 ankyrin 2 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2017-04-06.
- ↑ "KCNE1 potassium voltage-gated channel subfamily E regulatory subunit 1 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2017-04-06.
- ↑ "KCNE2 potassium voltage-gated channel subfamily E regulatory subunit 2 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2017-04-06.
- ↑ Hines, Roberta, Soeltin's Anesthesia and Co-existing Disease (4 ed.), Elsevir, p. 89
- ↑ "OMIM Entry - # 192500 - LONG QT SYNDROME 1; LQT1". omim.org. Retrieved 1 April 2017.
Further reading
- Kelly, Evelyn B. (2013-01-07). Encyclopedia of Human Genetics and Disease. ABC-CLIO. ISBN 9780313387135. retrieved 2017-04-07
- Nakano, Yukiko; Shimizu, Wataru (2016-01-01). "Genetics of long-QT syndrome". Journal of Human Genetics. 61 (1): 51–55. doi:10.1038/jhg.2015.74. ISSN 1434-5161.
External links
| Classification | |
|---|---|
| External resources |