Sikkelcelanemie
Sikkelcelanemie[2] en sikkelcelziekte zijn hemoglobinopathieën, ziekten aan het bloed waarbij in de rode bloedcellen het zuurstoftransporteiwit hemoglobine (Hb) veranderd is. Het zijn recessieve erfelijke aandoeningen: men erft het van beide ouders. Als iemand het gen van één ouder heeft gekregen (heterozygoot is), is diegene drager van het gen, maar heeft zelf meestal weinig klachten of symptomen. Als iemand het gen van beide ouders heeft gekregen (homozygoot is) dan heeft men doorgaans wel symptomen van sikkelcelziekte.
| Neem het voorbehoud bij medische informatie in acht. Raadpleeg bij gezondheidsklachten een arts. |
Sikkelcelanemie
| ||||
 | ||||
Gewone en sikkelvormige rode bloedcellen | ||||
| Synoniemen | ||||
| Latijn | anaemia drepanocytaria[1][2] | |||
| Nederlands | sikkelcellenanemie[1] drepanocytaire anemie[1] | |||
| Coderingen | ||||
| ICD-10 | D57 | |||
| ICD-9 | 282.6 | |||
| OMIM | 603903 | |||
| DiseasesDB | 1206 | |||
| MedlinePlus | 000527 | |||
| eMedicine | emerg/26 | |||
| MeSH | C15.378.071.141.150.150 | |||
| ||||
De meerderheid van deze populatie wordt door genetische selectie in balans gehouden, voornamelijk veroorzaakt door malaria en andere ziektes die normale rode bloedcellen aantasten en vernietigen, waardoor sikkelcelziekte als overlevingsaanpassing kan worden gezien.
Symptomen
Karakteristiek voor de ziekte is chronische hemolytische normocytaire anemie (bloedarmoede), episoden van pijnlijke botcrisen en de betrokkenheid van meerdere organen. Sikkelcelziekte gaat ook gepaard met een verhoogde stollingsactivatiestatus. Door de doorbloedingsstoornissen kunnen organen en weefsels beschadigingd raken (sikkelcelcrisis). Daarnaast hebben lijders aan de ziekte de neiging tot het oplopen van infecties, en kennen zij diverse andere complicaties.
Sikkelcel en mechanisme
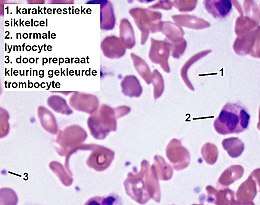
In plaats van de normale rode bloedcellen (erytrocyten) hemoglobine A (HbA, van 'adult') ontstaan sikkelcellen: abnormaal gevormde rode bloedcellen die hemoglobine S (HbS, van 'sickle') worden genoemd.
Dit wordt veroorzaakt door een puntmutatie van glutaminezuur naar valine op de β-keten van het hemoglobine-molecuul. Normaal hemoglobine bevat 4 polypeptideketens, waarvan 2 α-polypeptideketens en 2 β-polypeptideketens. β-polypeptideketens bevatten elk 146 aminozuurgroepen waarvan er bij sikkelcelanemie op positie 6 één vervangen is door valine. De sikkelcel (HbS) vormt spontaan polymeren die onoplosbaar zijn als de zuurstofspanning te ver daalt. Dit is het geval in kleine capillairen (haarvaten), waar de zuurstofconcentratie in (en buiten) de rode bloedcel laag is. Op de plekken waar het hemoglobine zijn zuurstof weer af staat, kristalliseert het, terwijl normaal Hb dat niet zou doen. Dit kan men nabootsen met behulp van een sikkelceltest. Als gevolg van deze afwijking krijgen de rode bloedcellen waarin het gepolymeriseerde HbS aanwezig is een sikkelachtige vorm en zijn ze ook veel minder soepel. Hierdoor worden de bloedcellen sneller afgebroken of klonteren ze soms spontaan samen of blijven steken in kleine capillairen (haarvaten), waardoor doorbloedingsstoornissen, ischemie en uiteindelijk eventueel necrose van organen en weefsels kunnen ontstaan (sikkelcelcrisis).
Types en terminologie
De meest voorkomende vorm van sikkelcelziekte is sikkelcelanemie. Bij die specifieke vorm is er een homozygositeit voor de mutatie die HbS veroorzaakt. Sikkelcelanemie wordt ook wel "HbSS", "sikkel-ziekte", of "hemoglobine S," (en alle permutaties ervan) genoemd.
Andere vormen van sikkelcelziekte zijn:
- sikkel-hemoglobine-C-ziekte
- sikkel β-plus-thalassemie
- sikkel β-0-thalassemie
Deze andere vormen van sikkelcelziekte zijn samengestelde heterozygote toestanden, waarin de persoon slechts één kopie van de mutatie heeft, die HbS veroorzaakt en één kopie van een andere abnormale hemoglobineallel. De term "ziekte" is hier toegepast omdat de abnormaliteit een pathologisch beeld veroorzaakt en tot de dood en ernstige complicaties kan leiden. Niet alle geërfde varianten van hemoglobine zijn schadelijk; een concept dat bekendstaat als genetisch polymorfisme.
Dragers van de ziekte
Het ziektebeeld komt vooral voor in etnische groepen uit landen waar malaria heerst. Dit betekent dat de ziekte meer voorkomt bij negroïde mensen of het nageslacht van negroïde mensen, vooral van West-Afrikaanse afkomst, maar ook vaker bij mensen van mediterrane afkomst, mensen uit het Midden-Oosten en van Indiase afkomst. Degenen met de heterozygote afwijking hebben evolutionair gezien een grotere overlevingskans in malariagebieden. Dit komt doordat de malariaparasiet, die zich in een bepaalde fase in rode bloedcellen vermenigvuldigt, zich in bloedcellen met hemoglobine S minder makkelijk kan handhaven. Er is enige tijd geleden dan ook een onderzoek gestart door een paar Nederlandse studenten die de afweer van sikkelcellen tegen malariaparasieten hebben vastgelegd en geanalyseerd. Het bleek een reactie te zijn van de malariaparasiet op de oplosbaarheid van het sikkelcelanemie-hemoglobine S. En het is dus ook moeilijk voor de parasiet om het hemoglobine tot zich te nemen als voedingsstof. Volgens onderzoek uit 2011 zijn sikkelcellen beschermd tegen malaria, omdat de parasiet het in deze cellen moeilijker heeft om actine van de cel te gebruiken voor zijn eigen levenscyclus.[3]
Onder normale omstandigheden komt de echte sikkelcelziekte alleen voor bij mensen die het afwijkende gen hebben geërfd van beide ouders. Dit is de zogenaamde homozygote vorm van de ziekte. In Amerika komt deze voor bij 1 op de 400 Afro-Amerikanen. Ongeveer 8% van de Afro-Amerikanen is heterozygoot voor de ziekte, dat wil zeggen dat zij slechts van één ouder het afwijkende gen hebben ontvangen. Zij zijn (meestal) symptoomloze dragers van de ziekte, maar kunnen wel symptomen krijgen indien zij ook lijden aan andere ernstige ziekten.
Behandeling van sikkelcelziekte en een crisis
Er is geen genezing mogelijk voor de ziekte, hoewel soms gentherapie als potentiële genezingswijze in de toekomst wordt genoemd. Moderne ontwikkelingen hebben het mogelijk gemaakt dat de gemiddelde levensduur (in het westen) is toegenomen tot 46 jaar.
De anemie wordt behandeld met foliumzuur. Vaak kan de ziekte ook behandeld worden door Hydrea (hydroxyureum oftewel hydroxycarbamide) te nemen, om aanmaak van niet-sikkelend hemoglobine te bevorderen en de kans op crises te verlagen.[4]
Sikkelcelcrises kunnen worden behandeld met:
- intraveneuze vochttoediening
- warmte
- pijnbestrijding
- zuurstof
- eventueel antibiotica bij (verdenking van) infecties
- eventueel bloedtransfusies bij extreem lage hemoglobine-gehaltes; vaak hebben deze mensen echter al meer bloedtransfusies gehad waardoor er vele antistoffen tegen verscheidene bloed-antigenen gemaakt worden. Hierdoor is het moeilijk een goede donor te vinden. Ook is er bij frequente transfusies gevaar voor ijzerstapeling.
- bij ernstige, terugkerende klachten kan een erytrocytaferese worden overwogen. De gesikkelde rode bloedcellen worden hierbij grotendeels vervangen door donorerytrocyten, waarmee tevens de kans op ijzerstapeling gereduceerd wordt. De erytrocytaferese kan met een bepaald interval (weken tot maanden) worden herhaald.
Organisaties
In Nederland
- oscarnederland.nl: Stichting Oscar Nederland, opgericht in 1987, met als doel meer bekendheid te geven aan sikkelcelziekte en thalassemie, voorlichting geven aan verschillende doelgroepen en patiënten en hun omgeving te ondersteunen.
- i-x-l.com: Stichting IXL ('I excel', Engels voor 'ik excelleer'), opgericht in 2015 met als doel meer bekendheid te geven aan sikkelcelziekte en patiënten en hun omgeving te ondersteunen.
Bronnen, noten en/of referenties
|