Sequentie-alignering
Sequentie-alignering (Engels: sequence alignment) is het bij elkaar zetten van twee of meer biologische sequenties op zo'n manier dat gelijkenissen en verschillen tussen de sequenties duidelijk worden. Dit soort alignering wordt binnen de biologie meestal aan computers overgelaten en vormt zo een bekend concept binnen de bio-informatica.
De sequenties, sequenties van DNA of eiwitten, worden hierbij gewoonlijk onder elkaar geplaatst. Gelijke onderdelen, respectievelijk nucleotiden en aminozuren, worden daarbij met verticale streepjes aan elkaar gelinkt.
Een voorbeeld voor de uitlijning van twee nucleotidensequenties:
tcctctgcctctgccatcat---caaccccaaagt |||| ||| ||||| ||||| |||||||||||| tcctgtgcatctgcaatcatgggcaaccccaaagt
Sequentie-alignering is van belang voor het nagaan van evolutionaire, structurele en functionele overeenkomsten in de sequenties van biologische moleculen. Zo kan verwantschap (homologie) van sequenties (door genverdubbeling (genduplicatie) of door soortvorming (speciatie) aangetoond worden.
Fouten (mismatches), gebruikelijk voorgesteld door het ontbreken van een verticaal streepje tussen twee elementen, kunnen verklaard worden door mutaties; gaten (gaps), voorgesteld door horizontale lijnen, door inserties of deleties. Die gaten worden ook wel "indels" genoemd, een lettergreepwoord dat komt van de termen "insertie" en "deletie".
Ook buiten de bio-informatica zijn toepassingen mogelijk, bijvoorbeeld bij de studie van taalverwantschap en tekstvergelijking.
Dynamische algoritmen
Dynamische algoritmes kunnen helpen bij het bepalen van de beste alignering tussen verschillende sequenties. De algoritmen maken gebruik van zogeheten substitutiematrices, waarin voor elke substitutie van een nucleotide/aminozuur in een ander (mismatch) en hetzelfde (match) een waarde staat. Er is ook een waarde voor een indel. Een match krijgt daarbij een gunstigere waarde dan een indel en mismatch. De meest plausibele alignering zal dan de alignering zijn met de gunstigste totale score. Een mismatch en indel doen de gelijkaardigheid tussen sequenties dalen, wat een minder gunstige score verklaart.
De uitlijning kan globaal of lokaal gebeuren. Bij globale uitlijning worden alle elementen van verwante sequenties in beschouwing genomen. Bij lokale uitlijning kunnen de beschouwde sequenties sterk ten opzichte van elkaar verschoven worden, zodat verschoven domeinen (domain shuffling) weer onder elkaar kunnen gezet worden. Bij een alignering van twee sequenties wordt daarbij bij elke sequentie een subset gezocht waarbij beide subsets een zo groot mogelijke globale alignering hebben. Er wordt dus op zoek gegaan naar een lokale gelijkenis, waarbij de rest van de sequentie niet meegerekend wordt en de score zo niet negatief beïnvloedt. Algemeen is een globaal algoritme het meest geschikt voor sequenties met matige tot veel gelijkenis, een lokaal is dan weer eerder geschikt voor sequenties met matige tot weinig gelijkenis. Vooral als er hele domeinen in een sequentie zijn omgewisseld of toegevoegd.
Globale alignering

Een veelgebruikt algoritme om twee volledige sequenties met elkaar te vergelijken, is het Needleman-Wunsch-algoritme. Daarbij worden twee sequenties in een matrixvorm geplaatst: de ene sequentie horizontaal, de andere verticaal, zoals in volgende tabel:
| C | A | C | G | A | ||
|---|---|---|---|---|---|---|
| 0 | ||||||
| C | ||||||
| A | ||||||
| T | ||||||
| T | ||||||
| G | ||||||
| A |
Van daaruit krijgt elk vakje in de matrix een bepaalde score. Er wordt gestart bij nul, waarna elk ander vakje zal worden opgevuld. Elke match, mismatch en indel krijgt een score volgens de substitutiematrix. Elk vakje krijgt nu een van volgende waarden:
- De waarde van het vak diagonaal linksboven vermeerderd met de substitutiewaarde van het vak zelf. Als het vak bijvoorbeeld een A (sequentie 1) tegenover een A (sequentie 2) plaatst, zal die waarde positief zijn. Een mismatch, een A tegenover een C bijvoorbeeld, zal dan weer minder goed scoren.
- De waarde van het vakje boven plus de waarde van een indel. Er wordt hierbij van uitgegaan dat om het vakje te bereiken een indel nodig is in de horizontale sequentie voor de beste alignering tussen de twee.
- De waarde van het linkse vakje plus de waarde van een indel. Idem als in het vorige puntje, maar dan een indel in de andere sequentie.
Het vakje zal steeds van deze drie mogelijkheden de waarde krijgen die de gunstigste score oplevert (afhankelijk van de gekozen matrix kan dat zowel het minimum als het maximum zijn). Een uitgebreidere versie, de oorspronkelijke versie, van dit algoritme houdt ook rekening met de lengte van indels. Bij twee of meer indels wordt dan niet de waarde van de indel gebruikt in de berekening, maar wel een functie die rekening houdt met het aantal indels na elkaar. Dit heeft ook een biologische waarde: het is aannemelijker dat indels in één keer plaatsvonden dan in afzonderlijke keren.

Uiteindelijk zal het vakje in de rechterbenedenhoek zo opgevuld worden. De sequentie wordt zo achterstevoren bepaald: de waarde van het vakje rechtsonder komt van een van de drie mogelijkheden. De bewuste mogelijkheid wordt dan bekeken: komt de waarde vanuit een substitutie (match of mismatch), dan is de gunstigste alignering diegene waarbij de laatste elementen tegenover elkaar staan in de alignering. Komt de waarde vanuit het vakje boven of links van het beschouwde, dan wordt een indel in de alignering geplaatst. In de volgende stap wordt dan naar dit vorige vakje gekeken, waarna de procedure zich herhaalt tot bovenaan links.
Lokale alignering
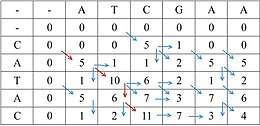
Het bekendste algoritme voor lokale alignering is een variant op het Needleman-Wunsch-algoritme: het Smith-Waterman-algoritme. Het grote verschil is dat hier geen "ongunstige" scores worden toegekend. Een indel en mismatch krijgen als waarde nul; alleen een match krijgt een gunstige waarde. De matrix wordt op dezelfde manier opgevuld, maar nu wordt gekeken naar de gunstigste waarde in de matrix. Van daaruit wordt dan op dezelfde manier teruggegaan naar de vorige stappen die de bewuste waarde hebben bepaald. Zo ontstaat uiteindelijk de lokale alignering.
Bij uitbreiding kunnen ook de stukken die buiten de alignering vallen nog eens onderworpen worden aan lokale aligneringen in een iteratief proces.
Substitutiematrixen
Bekende substitutiematrices voor aminozuren zijn gebaseerd op de PAM- en BLOSUM-matrix. Daarin wordt voor elk aminozuur een waarde toegekend voor de substitutie van dit aminozuur in een van de aminozuren. De waarde reflecteert de waarschijnlijkheid waarmee het aminozuur in een sequentie gesubstitueerd wordt door het tweede - niet per se verschillende - aminozuur.
PAM
De waarden in PAM-matrices zijn gebaseerd op vergelijkingen tussen sterk verwante aminozuurketens. PAM staat hierbij voor "point accepted mutation", een maat voor het verschil tussen sequenties. Eén PAM is daarbij de mate van verschil die optreedt als 1% van de aminozuren verandert. De waarde van een cel, stel , in de matrix staat voor de geobserveerde frequentie waarmee aminozuur vervangen werd door aminozuur . De PAM-matrix heeft bijvoorbeeld voor de cel voor R-R de waarde 0,9913, wat betekent dat 99,13% van de arginines (R) een arginine bleef in het geobserveerde geval waarbij 1% van de aminozuren werd gewijzigd. De cel voor R-N is dan weer 0,0001, wat betekent dat 1% van de arginines in asparagines (N) veranderde in hetzelfde geval.



De matrix waarbij 1% verandering wordt bekeken, krijgt de naam PAM-1. Door PAM-1 met zichzelf te vermenigvuldigen, wordt PAM-2 bekomen. Op eenzelfde manier kan dit veralgemeend worden tot PAM-. Bij bijvoorbeeld 250 PAM-eenheden wordt echter niet gesproken van 250% veranderde aminozuren; het betekent de mate van verandering die nodig is zodat elk aminozuur gemiddeld 2,5 keer werd gemuteerd.
Deze waarden kunnen vervolgens in een aligneringalgoritme worden gebruikt. De substitutiewaarde zal nu in functie zijn van de PAM-matrix (zie punt 1 bij de algoritmes). Er wordt hierbij dus niet één vaste waarde toegekend aan "een" match en "een" mismatch, maar er wordt gekeken naar de biologische achtergrond: sommige matchen en sommige mismatchen komen in de werkelijkheid meer voor dan andere, wat met PAM in rekening wordt gebracht. De nieuwe substitutiewaarde zal meer bepaald de volgende worden: . Hierbij staat voor de waarde in de gekozen PAM-matrix (PAM-1, PAM-2 ...) en is de relatieve frequentie van aminozuur .



Hoe minder de gealigneerde sequenties aan elkaar verwant zijn, hoe hoger de te verkiezen PAM-matrix.
BLOSUM
BLOSUM is een letterwoord dat staat voor blocks substitution matrix. De gebruikte waarden in de matrix zijn gebaseerd op een meervoudige alignering (alignering van een aantal sequenties samen) van geconserveerde sequenties (sequenties die over generaties en soorten heen slechts licht variëren). De naam van BLOSUM-matrix wordt net als bij PAM gevolgd door een nummer. BLOSUM 62 bijvoorbeeld betekent daarbij dat de waarden bepaald zijn aan de hand van sequenties die 62% gelijk zijn. Hoe minder verwant de vergeleken sequenties, des te lager de te gebruiken BLOSUM-matrix; net het omgekeerde van bij PAM.
Heuristieken
De Needleman-Wunsch- en Smith-Waterman-algoritmes kunnen op praktische problemen stuiten. Ze leveren wel de optimale alignering voor het gegeven scoresysteem, maar de complexiteit ervan is relatief hoog. Ze hebben een complexiteit van de grootteorde , omdat telkens een matrix van grootte op moet worden opgevuld. In de praktijk kunnen die waarden duizenden tekens lang zijn. Dit is rekenintensief, waardoor de uitvoering computationeel (te) moeilijk is, en zal vooral praktische problemen vertonen als een sequentie vergeleken wordt met veel andere sequenties. Om die rekenintensiviteit te verkleinen, wordt vaak gebruikgemaakt van heuristieken. Daarmee wordt het zoekproces drastisch verkleind, met weliswaar als risico de beste alignering te mislopen. Met een goede heuristiek is dat risico beperkt en in de praktijk verwaarloosbaar. Bekende heuristische algoritmes zijn FASTA en BLAST. Zij maken niet alleen een alignering, maar kennen ook een waarde voor statische toepasselijkheid aan de alignering toe. Er blijft echter een zekere kans op fout-positieven bestaan: het vinden van een homologie waar die er niet echt is.


FASTA
FASTA verdeelt de sequenties in zogeheten -tuples, waarbij een natuurlijk getal is. De sequenties worden opgesplitst in groepjes van elementen (letters). Elke mogelijke combinatie krijgt daarbij een unieke waarde. Elke gevormde tuple krijgt ook een volgnummer, dat zijn plaats in de sequentie definieert. Vervolgens worden de sequenties vergeleken. Bij waarden die overeenkomen, wordt gekeken naar het verschil in volgnummer. Verschillen die vaak voorkomen, wijzen op gelijkenissen tussen de sequenties. De grootste gebieden met gelijkenissen worden vervolgens aan elkaar gelinkt met behulp van een scoresysteem met een substitutiematrix en het toestaan van gaten tussen de gelinkte gebieden.

De complexiteit van het algoritme blijft in de meest voorkomende gevallen van de grootteorde .

BLAST
FASTA werkt met exacte overeenkomsten tussen de -tuples, BLAST is hier echter iets flexibeler in. Niet alleen exacte overeenkomsten worden in rekening gebracht, maar ook gelijkenissen die binnen bepaalde te kiezen grenzen blijven. De gelijkenissen tussen de tuples krijgen met behulp van een substitutiematrix een waarde. Als die waarde beter is dan de vooropgestelde grenswaarde, dan worden de tuples in het verdere proces meegenomen.
BLAST past eerst de heuristiek toe om vervolgens slechts met een deel van het originele probleem verder te moeten werken. Ook bij BLAST bevindt de complexiteit zich in de grootteorde .
Meervoudige alignering
Aligneren beperkt zich niet tot het paarsgewijs vergelijken, dus van twee sequenties, maar kan ook toegepast worden op het tegelijk aligneren van meerdere sequenties. Het achterliggende idee blijft hetzelfde: er wordt gezocht hoe de sequenties onder elkaar kunnen woorden voorgesteld zodat hun verwantschap duidelijk wordt. Net als bij paarsgewijze alignering kan er aan de alignering een beoordelende waarde toegekend worden. Een manier om elke gealigneerde kolom te beoordelen, is het onderling vergelijken van elke sequentie met elke andere voor de bewuste kolompositie en dit op te tellen. Dit betekent dat (een wiskundige combinatie) paren worden vergeleken, waarbij staat voor het aantal sequenties.

De aligneringen zelf kunnen worden gemaakt met uitbreidingen van de algoritmen voor paarsgewijze alignering (zowel de heuristische als de andere). In plaats van met twee dimensies te werken, wordt dan met dimensies gewerkt. De complexiteit zal echter ook mee stijgen. Voor het Smith-Waterman-algoritme zal voor elk element in de matrix een berekening van de grootteorde nodig zijn. Voor de hele matrix betekent dat een complexiteit van grootteorde , waarbij staat voor de lengte van de sequenties.


In realistische situaties is dit te complex voor nuttig gebruik. In de praktijk wordt daarom vaak voor een andere aanpak gekozen: het zoeken naar consensussequenties. Daarbij wordt gezocht naar de twee meest gelijkaardige sequenties en wordt daarvan een consensus opgesteld. Die zal de twee sequenties vervangen, waarna de procedure herhaald wordt, met de consensus als een van de te beschouwen sequenties. Door na de laatste alignering de consensussequenties weer uiteen te halen, wordt de alignering bekomen. Tijdens het zoeken wordt in feite een boomstructuur opgesteld, die de guide tree genoemd wordt, een benadering van een fylogenetische boom. De bladen van deze structuur zijn dan de sequenties, de andere knopen zijn de gevormde consensussen. De wortel van die boom is dan de consensus van alle sequenties. De meervoudige alignering wordt dan aan de hand van die gevormde boom opgesteld. Algoritmes die dit principe gebruiken, kunnen verschillen in de manier waarop consensussen bepaald worden en de manier waarop de boom opgebouwd wordt.
Bij het zoeken naar consensussequenties worden zo'n paarsgewijze aligneringen uitgevoerd, wat als bovengrens heeft. Dit vermenigvuldigen met de grootteorde waarmee een paarsgewijze alginering wordt uitgevoerd, bepaalt de totale grootteorde. Voor BLAST of FASTA wordt dit een orde in de buurt van . Een nadeel van deze methode is dat aligneringsfouten in een beginstadium worden meegenomen naar de volgende stadia. Er bestaan algoritmes die dit probleem proberen aan te pakken met iteratieve aligneringalgoritmes, zodat fouten in een eerdere stap worden rechtgezet. Het is echter niet steeds zo dat dit convergeert, waardoor het nuttig is om vooraf in te stellen hoeveel iteraties maximaal mogen worden uitgevoerd.


Het zoeken van consensussequenties gebeurt door het zoeken naar de waarschijnlijkste evolutionaire verandering tussen twee vergeleken sequenties en dat laten passen binnen de gehele alignering. Daarbij moet gezocht worden naar een compromis tussen die twee.
Meervoudige alginering wordt vooral gebruikt om structurele of functionele verbanden te zoeken tussen sequenties en sequentiefamilies. Zo kunnen variabele en geconserveerde plaatsen in eiwitfamilies gevonden worden en het kan informatie bieden over evolutionaire geschiedenis.
Een bekend programma om meervoudige alignering mee uit te voeren, is Clustal.
Externe links
- Blast Server van het NCBI
- Tutorial over BLAST-algoritme en de statistische significantie van sequentie-uitlijningen in het algemeen
- JAligner: opensource-Java-programma van dynamisch programmerings-algoritme Smith-Waterman voor paarsgewijze uitlijning
Bron
|