Machado–Joseph disease
| Machado-Joseph disease | |
|---|---|
| Synonyms | Autosomal dominant striatonigral degeneration, Nigro-spino-dentatal degeneration with nuclear ophthalmoplegia |
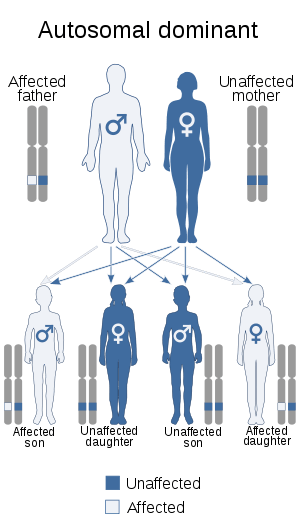 | |
| Machado–Joseph disease is inherited via an autosomal dominant manner | |
| Specialty |
Neurology |
Machado–Joseph disease (MJD), also known as Machado–Joseph Azorean disease, Machado's disease, Joseph's disease or spinocerebellar ataxia type 3 (SCA3), is a rare autosomal dominantly inherited neurodegenerative disease that causes progressive cerebellar ataxia,[1][2] which results in a lack of muscle control and coordination of the upper and lower extremities.[3] The symptoms are caused by a genetic mutation that results in an expansion of abnormal "CAG" trinucleotide repeats in the ATXN3 gene [1] that results in an abnormal form of the protein ataxin which causes degeneration of cells in the hindbrain.[3] Some symptoms, such as clumsiness and rigidity, make MJD commonly mistaken for drunkenness or Parkinson's disease.
Machado–Joseph disease is a type of spinocerebellar ataxia and is the most common cause of autosomal-dominant ataxia.[1] MJD causes ophthalmoplegia and mixed sensory and cerebellar ataxia.
Symptoms
Symptoms of MJD are memory deficits,[4] spasticity, difficulty with speech and swallowing, weakness in arms and legs, clumsiness, frequent urination and involuntary eye movements. Symptoms can begin in early adolescence and they get worse over time. Eventually, MJD leads to paralysis; however, intellectual functions usually remain the same.
Pathophysiology
The disease is caused by a mutation in the ATXN3 gene, which is located on chromosome 14q. The gene contains lengthy irregular repetitions of the code "CAG", producing a mutated protein called ataxin-3. (Normally, the number of copies is between 13 and 41.)[5] MJD is an autosomal dominant disease, meaning that if either parent gives the defective gene to a child, the child will show symptoms of the disease. Therefore, if one parent suffers from this disease and the other parent does not, there will be a 50% chance of their child inheriting the disease.[3]
The pons (a structure located on the brain stem) is one of the areas affected by MJD. The striatum (a brain area connected to balance and movement) is also affected by this disease, which could explain both of the main motor problems cause by MJD: the tightening and twisting of the limb and the abrupt, irregular movements.[6]
In affected cells, this protein builds up and assembles intranuclear inclusion bodies. These insoluble aggregates are hypothesized to interfere with the normal activity of the nucleus and induce the cell to degenerate and die.
Diagnosis
MJD can be diagnosed by recognizing the symptoms of the disease and by taking a family history. Physicians ask patients questions about the kind of symptoms relatives with the disease had, the progression and harshness of symptoms, and the ages of onset in family members.
Presymptomatic diagnosis of MJD can be made with a genetic test.[7] The direct detection of the genetic mutation responsible for MJD has been available since 1995.[8] Genetic testing looks at the number of CAG repeats within the coding region of the MJD/ATXN3 gene on chromosome 14. The test will show positive for MJD if this region contains 61-87 repeats, as opposed to the 12-44 repeats found in healthy individuals. A limitation to this test is that if the number of CAG repeats in an individual being tested falls between the healthy and pathogenic ranges (45-60 repeats), then the test cannot predict whether an individual will have MJD symptoms.[7]
Classification
There are five sub-types of MJD [1] that are characterized by the age of onset and range of symptoms. The sub-types illustrate a wide variety of symptoms that patients can experience.[1] However, assigning individuals to a specific sub-type of the disease is of limited clinical significance.[1]
- Type I is distinguished by arrival between the ages of 10 and 30 and represents approximately 13% of individuals.[1] It usually has fast development and severe rigidity and dystonia.
- Type II is the most common sub-type (approximately 57% of individuals with MJD [1]) and typically begins between 20 and 50 years of age . It has an intermediate progression and causes symptoms that include spasticity, exaggerated reflex responses and spastic gait, ataxia [1] and upper motor neuron signs.[1]
- Type III MJD has a slow progression. Patients typically have an onset between the ages of 40 and 70 and represent approximately 30% of MJD patients.[1] Symptoms include muscle twitching, tingling, cramps, unpleasant sensations such as numbness, pain in the feet, hands and limbs and muscle atrophy. Nearly all patients experience a decline in their vision such as blurred vision, double vision, inability to control eye movements, and loss of capability to distinguish color. Some patients also experience Parkinsonian symptoms.
- Type IV is distinguished by Parkinsonian symptoms that respond particularly well to levodopa treatment.[1]
- Type V appears to resemble Hereditary Spastic Paraplegia; however, more research is needed to conclude the relationship between Type V MJD and hereditary spastic paraplegia.[1]
Treatment
There is no cure for Machado-Joseph Disease. However, treatments are available for some symptoms.[1][3] For example, spasticity can be reduced with antispasmodic drugs, such as baclofen. The Parkinsonian symptoms can be treated with levodopa therapy. Prism glasses can reduce diplopic symptoms.[3] Physiotherapy/Physical Therapy and/or occupational therapy can help patients by prescribing mobility aids to increase the patients' independence, providing gait training, and prescribing exercises to maintain the mobility of various joints and general health to decrease the likelihood of falls or injuries as a result of falls. Walkers and wheelchairs can greatly help the patient with everyday tasks. Some patients will experience difficulties with speech and swallowing, therefore a Speech-Language Pathologist can assist the patients to improve their communicating abilities and their issues with swallowing.[3]
Prognosis
Patients with severe forms of MJD have a life expectancy of approximately 35 years. Those with mild forms have a normal life expectancy. The cause of death of those who die early is often aspiration pneumonia.[3]
History
The disease was first identified in 1972.[9]
Unlike many other medical conditions, Machado–Joseph disease is not named after researchers. It is named after two men ("William Machado" and "Antone Joseph") who were the patriarchs of the families in which the condition was initially described.[10] The highest prevalence of the condition is on Australia's Groote Eylandt where 5% of the population are currently symptomatic or at risk,[11] followed by the Azorean island of Flores where around 1 in 140 individuals in the population are diagnosed with MJD.[12]
Flores and São Miguel are centers of the Machado–Joseph disease in the Azores.[13]
Machado Joseph's disease has multiple origins as SCA3 comes from haplotype of four different origins and was not from one origin in the Azores. Japan, Brazil and France all have been founder effects in areas with SCA3.[14]
Spinocerebellar ataxia type 3 (SCA3) on the Azores are believed to have come from Portugal's northeast where Sephardic Jews lived.[15] Belgium, French-Guiana, and Algeria have their own MJD mutation origins while Portuguese have two mutations, while MJD in Brazil and France have one origin, and Germans make up the majority of MHD patients in the United States. Azorean MJD suffers have their locus on the 14q24.3-32 chromosome, the same as some Japanese with MJD. It was an Azorean William Machado, whose offspring in New England were first to be diagnosed with MJD.[16][17] The Azorean Californian Joseph family had MJD besides the Azorean Machados. The same origin for MJD is found in the Azores and in America's north-west coast. Not only Portuguese have it since African Americans, Indians, Italians and Japanese also developed MJD.[18]
MJD affected Azroeans and Japanese had haplotypes in common while Azoreans also had haplotypes of two different origins.[19]
Notable cases
Brazilian comedian, actor and TV personality Guilherme Karam was diagnosed with Machado–Joseph disease, having inherited it from his mother, like his brother and sister.[20] He died on 7 July 2016.[21] In a video on Facebook just days after Karam's death, Brazilian personality, journalist and television presenter, Arnaldo Duran publicly acknowledged his affliction with Machado-Joseph. Brazilian press gave huge attention for both cases.[22]
Ethical consideration
Ethicists have used Machado–Joseph disease as a paradigmatic illness to discuss the rights of a community of patients to control "ownership" of their disease, particularly when it comes to research on genetic testing.[23] Also, as there currently is no clinical intervention to prevent the onset of the disease symptoms, there is discourse over whether individuals should get tested or not.[8] The benefits of having MJD testing include a reduction in anxiety and uncertainty, and the ability to plan for the future. Some disadvantages include the anticipation of negative results and the individual's difficulties in adapting to this outcome.
For an ethnographic case study exploring some of the social and ethical consequences of living with Machado–Joseph disease, see João Biehl's Vita: Life in a Zone of Social Abandonment (Berkeley: University of California Press, 2006).
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Paulson, H. (8 March 2011). Spinocerebellar Ataxia Type 3. In R. A. Pagon, T. D. Bird, C. R. Dolan, & K. Stephens (Eds.), GeneReviews. Seattle, WA: University of Washington. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1196/
- ↑ Bettencourt, Conceição; Lima, Manuela (2011). "Machado-Joseph Disease: from first descriptions to new perspectives". Orphanet Journal of Rare Diseases. 6: 35. doi:10.1186/1750-1172-6-35. PMC 3123549. PMID 21635785. Retrieved 12 April 2015.
- 1 2 3 4 5 6 7 "Machado-Joseph Disease Fact Sheet". Retrieved 14 October 2016.
- ↑ Kawai Y, Takeda A, Abe Y, Washimi Y, Tanaka F, Sobue G (November 2004). "Cognitive impairments in Machado-Joseph disease". Arch. Neurol. 61 (11): 1757–60. doi:10.1001/archneur.61.11.1757. PMID 15534186.
- ↑ Lima M, Costa MC, Montiel R, et al. (2005). "Population genetics of wild-type CAG repeats in the Machado-Joseph disease gene in Portugal". Hum. Hered. 60 (3): 156–63. doi:10.1159/000090035. PMID 16340213.
- ↑ "cienciahoje.pt". Retrieved 14 October 2016.
- 1 2 Maciel P, Costa MC, Ferro A, et al. (November 2001). "Improvement in the molecular diagnosis of Machado-Joseph disease". Arch. Neurol. 58 (11): 1821–7. doi:10.1001/archneur.58.11.1821. PMID 11708990.
- 1 2 Rolim L, Zagalo-Cardoso JA, Paul C, Sequeiros J, Fleming M (2006). "The perceived advantages and disadvantages of presymptomatic testing for Machado-Joseph Disease: development of a new self-response inventory". Journal of Genetic Counseling. 15 (5): 375–391. doi:10.1007/s10897-006-9033-8.
- ↑ Nakano KK, Dawson DM, Spence A (1972). "Machado disease. A hereditary ataxia in Portuguese emigrants to Massachusetts". Neurology. 22 (1): 49–55. doi:10.1212/wnl.22.1.49. PMID 5061839.
- ↑ "SCA-3 - Ataxia Center in the Medical School at the University of Minnesota".
- ↑ "Paralysis island: 'slowly the disease kills them'". Retrieved 2016-11-14.
- ↑ "Machado-Joseph Disease Fact Sheet". Retrieved 2011-05-09.
- ↑ Gaspar, C.; Lopes-Cendes, I.; Hayes, S.; Goto, J.; Arvidsson, K.; Dias, A.; Silveira, I.; Maciel, P.; Coutinho, P.; Lima, M.; Zhou, Y.-X.; Soong, B.-W.; Watanabe, M.; Giunti, P.; Stevanin, G.; Riess, O.; Sasaki, H.; Hsieh, M.; Nicholson, G. A.; Brunt, E.; Higgins, J. J.; Lauritzen, M.; Tranebjaerg, L.; Volpini, V.; Wood, N.; Ranum, L.; Tsuji, S.; Brice, A.; Sequeiros, J.; Rouleau, G. A.; et al. (February 2001). "Ancestral Origins of the Machado-Joseph Disease Mutation: A Worldwide Haplotype Study". American Journal of Human Genetics. 68 (2): 523–528. doi:10.1086/318184. PMC 1235286. PMID 11133357.
- ↑ Puneet Opal; Henry Paulson (2000). Marie-Francoise Chesselet, ed. Molecular Mechanisms of Neurodegenerative Diseases (illustrated ed.). Springer Science & Business Media. p. 286. ISBN 1592590063. Retrieved 2014-02-02.
- ↑ Thomas Klockgether (2000). Handbook of Ataxia Disorders. CRC Press. p. 387. ISBN 1420002066. Retrieved 2014-02-02.
- ↑ David S. Younger. David S. Younger, ed. Motor Disorders. p. 663. Retrieved 2014-02-02.
- ↑ Paul Goodman (2004). Software Metrics: Best Practices for Successful IT Management (illustrated ed.). Rothstein Associates Inc. ISBN 1931332266. Retrieved 2014-02-02.
- ↑ Noshir H. Wadia (2005). Neurological Practice: An Indian Perspective. Elsevier India. p. 424. ISBN 8181475496. Retrieved 2014-02-02.
- ↑ Stefan M. Pulst (2002). Genetics of Movement Disorders. Academic Press. p. 63. ISBN 0080532411. Retrieved 2014-02-02.
- ↑ "'Ele perdeu a alegria de viver', diz pai de Guilherme Karam, que sofre de uma doença degenerativa". Retrieved 14 October 2016.
- ↑ "Morre o ator Guilherme Karam, aos 58 anos - Cultura - Estadão". Retrieved 14 October 2016.
- ↑ "Arnaldo Duran revela que está com doença neurológica degenerativa, sem cura e fatal - Entretenimento - R7 Famosos e TV". Retrieved 14 October 2016.
- ↑ Appel J, Friedman JH (January 2004). "Genetic markers and the majority's right not to know". Mov. Disord. 19 (1): 113–4. doi:10.1002/mds.20014. PMID 14743372.
External links
| Classification |
|---|