Tyrosinemia
| Tyrosinemia | |
|---|---|
 | |
| Tyrosine | |
| Specialty |
Endocrinology |
Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms include liver and kidney disturbances and intellectual disability. Untreated, tyrosinemia can be fatal.[1] Most inborn forms of tyrosinemia produce hypertyrosinemia (high levels of tyrosine).[2]
Cause
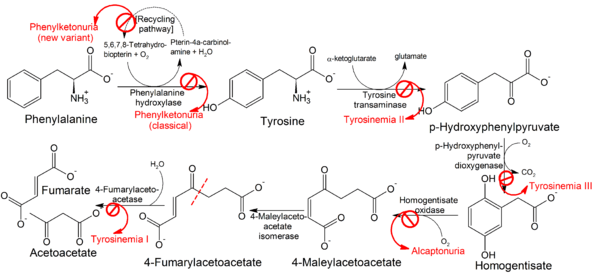
All tyrosinemias result from dysfunction of various genes in the phenylalanine and tyrosine catabolic pathway, and are inherited in an autosomal-recessive pattern.[3]
Type I tyrosinemia results from a mutation in the FAH gene, which encodes the enzyme fumarylacetoacetase.[4] As a result of FAH deficiency, the substrate fumarylacetoacetate can accumulate in proximal renal tubular cells and hepatocytes, resulting in damage to the kidney and liver, respectively.[3]
Type II tyrosinemia results from a mutation in the TAT gene, which encodes the enzyme tyrosine aminotransferase.[4] As a result of TAT deficiency, the substrate tyrosine accumulates, causing ophthalmologic and dermatologic abnormalities.[3]
Type III tyrosinemia results from a mutation in the HPD gene, which encodes the enzyme 4-hydroxyphenylpyruvate dioxygenase.[4] Type III tyrosinemia is the rarest of the three conditions, with only a few cases ever reported.[5] Most of those cases have included intellectual disability and neurologic dysfunction.[3]
Diagnosis
Types
Type I tyrosinemia can be detected via blood tests for the presence of a fumarylacetoacetate metabolite, succinylacetone, which is considered a pathognomonic indicator for the disease.[6]
Type II tyrosinemia can be detected via the presence of significantly elevated plasma tyrosine levels, and the diagnosis can be confirmed by detection of a mutation in TAT in cultured fibroblasts.
Type III tyrosinemia can be diagnosed by detection of a mutation in HPD in cultured fibroblasts.[3]
Treatment
Treatment varies depending on the specific type; a low-protein diet may be required. Recent experience with nitisinone has shown it to be effective. It is a 4-hydroxyphenylpyruvate dioxygenase inhibitor indicated for the treatment of hereditary tyrosinemia type 1 (HT-1) in combination with dietary restriction of tyrosine and phenylalanine.[7] Liver transplant is indicated for patients with tyrosinemia type I who do not respond to nitisinone, as well as those with acute liver failure and hepatomas.[8]
See also
References
- ↑ Shaw, Kathy; Bachur, Richard (2016). Fleisher & Ludwig's Textbook of Pediatric Emergency Medicine. Wolters Kluwer. ISBN 978-1451193954.
- ↑ Charles Scriver, Beaudet, A.L., Valle, D., Sly, W.S., Vogelstein, B., Childs, B., Kinzler, K.W. (Accessed 2007). The Online Metabolic and Molecular Bases of Inherited Disease. Chapter 79. New York: McGraw-Hill.
- 1 2 3 4 5 Grompe, Markus (2016-12-20). "Disorders of Tyrosine Metabolism". www.uptodate.com. Retrieved 2018-02-23.
- 1 2 3 Nelson, David; Cox, Michael (2013). Lehninger Principles of Biochemistry (6th. ed.). New York: WH Freeman and Co. p. 719. ISBN 978-1-4292-3414-6.
- ↑ "Tyrosinemia Type III detected via neonatal screening: Management and outcome". Molecular Genetics and Metabolism. 107 (3): 605–607. 2012-11-01. doi:10.1016/j.ymgme.2012.09.002. ISSN 1096-7192 – via Elsevier Science Direct.
- ↑ "Succinylacetone as primary marker to detect tyrosinemia type I in newborns and its measurement by newborn screening programs". Molecular Genetics and Metabolism. 113 (1–2): 67–75. 2014-09-01. doi:10.1016/j.ymgme.2014.07.010. ISSN 1096-7192. PMC 4533100 – via Elsevier Science Direct.
- ↑ Swedish Orphan Biovitrum AB, Orfadin [package insert] (PDF), retrieved 2016-07-12
- ↑ Mieles, L.A.; Esquivel, C.O.; Van Thiel, D.H.; Koneru, B.; Makowka, L.; Tzakis, A.G.; Starzl, T.E. (January 1990). "Liver Transplantation for Tyrosinemia". Digestive Diseases and Sciences. 35 (1): 153–157. PMC 2974306. PMID 2153069.
External links
| Classification | |
|---|---|
| External resources |
- GeneReview/NCBI/NIH/UW entry on Tyrosinemia Type 1
- Tyrosinemia on Genetic Home Reference