Argininemia
| Argininemia | |
|---|---|
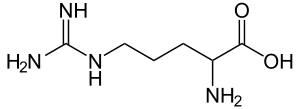 | |
| Arginine | |
| Specialty |
Neurology, Medical genetics, Endocrinology |
| Symptoms | Lethargy, Dehydration[1][2] |
| Causes | Mutations in the ARG1 gene[3][4] |
| Diagnostic method | Urinary orotic acid concentration[1] |
| Treatment | Protein intake limited, Sodium benzoate[2] |
Argininemia, also called arginase deficiency,[5] is an autosomal recessive urea cycle disorder where a deficiency of the enzyme arginase causes a buildup of arginine and ammonia in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if levels become too high; the nervous system is especially sensitive to the effects of excess ammonia.[1][6]
Signs/symptoms
The presentation of argininemia, in those that are affected, is consistent with the following:[1][2]
- Lethargy
- Dehydration
- Hypotonia
- Growth is stunted
- Microcephaly
- Seizures
Genetics
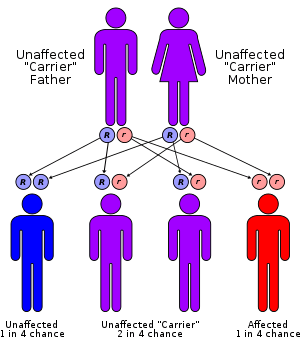
Mutations in the ARG1 gene cause argininemia, which belongs to a class of genetic diseases called urea cycle disorders.[3][4] The urea cycle is a sequence of reactions that occurs in liver cells(hepatocytes). This cycle processes excess nitrogen, generated when protein is used by the body, making urea that is excreted via the kidneys.[7]
The ARG1 gene provides instructions for making an enzyme called arginase, this enzyme controls the last steps of the urea cycle, which produces urea by extracting nitrogen from arginine.[3] In people with arginase deficiency, arginase is missing, and arginine is not broken down properly. consequently, urea cannot be produced and excess nitrogen accumulates in the blood in the form of ammonia. Ammonia and arginine are thought to cause neurological problems and other symptoms of arginase deficiency.[1]
This condition is an autosomal recessive disorder, which means the defective gene is located on an autosome,[6] and two copies of the defective gene are required to inherit the disorder. Both parents of an individual with an autosomal recessive disorder are carriers of one copy of the gene, but usually do not have the disorder.
Diagnosis
The diagnosis for argininemia can usually be done using fetal blood sample.[8] One can look for the following indicators as to the presence of the condition:[1]
- Plasma ammonia concentration.
- Urinary orotic acid concentration
- Red blood cell arginase enzyme activity(measurement)
Treatment

The treatment for infants (individuals) with argininemia is the following, including medications:[2]
References
- 1 2 3 4 5 6 Wong, Derek; Cederbaum, Stephen; Crombez, Eric A. (1 January 1993). "Arginase Deficiency". GeneReviews(®). University of Washington, Seattle. Retrieved 20 November 2016. update 2014
- 1 2 3 4 "Arginase Deficiency: Background, Pathophysiology, Epidemiology". eMedicine. 15 April 2016. Retrieved 28 November 2016.
- 1 2 3 Reference, Genetics Home. "ARG1 gene". Genetics Home Reference. Retrieved 28 November 2016.
- 1 2 Ah Mew, Nicholas; Lanpher, Brendan C.; Gropman, Andrea; Chapman, Kimberly A.; Simpson, Kara L.; Summar, Marshall L. (1 January 1993). "Urea Cycle Disorders Overview". GeneReviews(®). University of Washington, Seattle. Retrieved 20 November 2016. update 2015
- ↑ Online Mendelian Inheritance in Man (OMIM) 207800
- 1 2 Reference, Genetics Home. "arginase deficiency". Genetics Home Reference. Retrieved 20 November 2016.
- ↑ Hames, David; Hooper, Nigel (2005). Instant Notes in Biochemistry (3rd ed.). Hoboken: Taylor & Francis Ltd. p. 408. ISBN 9780203967621. Retrieved 28 November 2016.
- ↑ Wyllie, Robert; Hyams, Jeffrey S.; Kay, Marsha (2015). Pediatric Gastrointestinal and Liver Disease. Elsevier Health Sciences. p. 886. ISBN 9780323370219.
Further reading
- (editors), Jean-Marie Saudubray, Georges van den Berghe, John H. Walter; Berghe, Georges van den; Walter, John H. (2012). Inborn metabolic diseases diagnosis and treatment (5th ed.). Berlin: Springer. ISBN 9783642157202. Retrieved 28 November 2016.
- Piña-Garza, J. Eric (2013). Fenichel's Clinical pediatric neurology a signs and symptoms approach (7th ed.). Oxford: Saunders. ISBN 1455748129. Retrieved 28 November 2016.
External links
| Classification | |
|---|---|
| External resources |