Mutation
In biology, a mutation is an alteration in the nucleotide sequence of the genome of an organism, virus, or extrachromosomal DNA.[1] Viral genomes can be of either DNA or RNA. Mutations result from errors during DNA replication, mitosis, and meiosis or other types of damage to DNA (such as pyrimidine dimers that may be caused by exposure to radiation or carcinogens), which then may undergo error-prone repair (especially microhomology-mediated end joining[2]) or cause an error during other forms of repair[3][4] or else may cause an error during replication (translesion synthesis). Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.[5][6][7]
| Part of a series on |
| Evolutionary biology |
|---|
 Darwin's finches by John Gould |
|
Processes and outcomes
|
|
Natural history
|
|
History of evolutionary theory
|
|
Fields and applications
|
|
Social implications
|
|
| Part of a series on |
| Genetics |
|---|
 |
| Key components |
|
| History and topics |
|
| Research |
|
|
| Personalized medicine |
| Personalized medicine |

Mutations may or may not produce discernible changes in the observable characteristics (phenotype) of an organism. Mutations play a part in both normal and abnormal biological processes including: evolution, cancer, and the development of the immune system, including junctional diversity.
Mutation can result in many different types of change in sequences. Mutations in genes can either have no effect, alter the product of a gene, or prevent the gene from functioning properly or completely. Mutations can also occur in nongenic regions. A 2007 study on genetic variations between different species of Drosophila suggested that, if a mutation changes a protein produced by a gene, the result is likely to be harmful, with an estimated 70% of amino acid polymorphisms that have damaging effects, and the remainder being either neutral or marginally beneficial.[8] Due to the damaging effects that mutations can have on genes, organisms have mechanisms such as DNA repair to prevent or correct mutations by reverting the mutated sequence back to its original state.[5]
Overview
Mutations can involve the duplication of large sections of DNA, usually through genetic recombination.[9] These duplications are a major source of raw material for evolving new genes, with tens to hundreds of genes duplicated in animal genomes every million years.[10] Most genes belong to larger gene families of shared ancestry, detectable by their sequence homology.[11] Novel genes are produced by several methods, commonly through the duplication and mutation of an ancestral gene, or by recombining parts of different genes to form new combinations with new functions.[12][13]
Here, protein domains act as modules, each with a particular and independent function, that can be mixed together to produce genes encoding new proteins with novel properties.[14] For example, the human eye uses four genes to make structures that sense light: three for cone cell or color vision and one for rod cell or night vision; all four arose from a single ancestral gene.[15] Another advantage of duplicating a gene (or even an entire genome) is that this increases engineering redundancy; this allows one gene in the pair to acquire a new function while the other copy performs the original function.[16][17] Other types of mutation occasionally create new genes from previously noncoding DNA.[18][19]
Changes in chromosome number may involve even larger mutations, where segments of the DNA within chromosomes break and then rearrange. For example, in the Homininae, two chromosomes fused to produce human chromosome 2; this fusion did not occur in the lineage of the other apes, and they retain these separate chromosomes.[20] In evolution, the most important role of such chromosomal rearrangements may be to accelerate the divergence of a population into new species by making populations less likely to interbreed, thereby preserving genetic differences between these populations.[21]
Sequences of DNA that can move about the genome, such as transposons, make up a major fraction of the genetic material of plants and animals, and may have been important in the evolution of genomes.[22] For example, more than a million copies of the Alu sequence are present in the human genome, and these sequences have now been recruited to perform functions such as regulating gene expression.[23] Another effect of these mobile DNA sequences is that when they move within a genome, they can mutate or delete existing genes and thereby produce genetic diversity.[6]
Nonlethal mutations accumulate within the gene pool and increase the amount of genetic variation.[24] The abundance of some genetic changes within the gene pool can be reduced by natural selection, while other "more favorable" mutations may accumulate and result in adaptive changes.

For example, a butterfly may produce offspring with new mutations. The majority of these mutations will have no effect; but one might change the color of one of the butterfly's offspring, making it harder (or easier) for predators to see. If this color change is advantageous, the chances of this butterfly's surviving and producing its own offspring are a little better, and over time the number of butterflies with this mutation may form a larger percentage of the population.
Neutral mutations are defined as mutations whose effects do not influence the fitness of an individual. These can increase in frequency over time due to genetic drift. It is believed that the overwhelming majority of mutations have no significant effect on an organism's fitness.[25][26] Also, DNA repair mechanisms are able to mend most changes before they become permanent mutations, and many organisms have mechanisms for eliminating otherwise-permanently mutated somatic cells.
Beneficial mutations can improve reproductive success.[27][28]
Causes
Four classes of mutations are (1) spontaneous mutations (molecular decay), (2) mutations due to error-prone replication bypass of naturally occurring DNA damage (also called error-prone translesion synthesis), (3) errors introduced during DNA repair, and (4) induced mutations caused by mutagens. Scientists may also deliberately introduce mutant sequences through DNA manipulation for the sake of scientific experimentation.
One 2017 study claimed that 66% of cancer-causing mutations are random, 29% are due to the environment (the studied population spanned 69 countries), and 5% are inherited.[29]
Humans on average pass 60 new mutations to their children but fathers pass more mutations depending on their age with every year adding two new mutations to a child.[30]
Spontaneous mutation
Spontaneous mutations occur with non-zero probability even given a healthy, uncontaminated cell. They can be characterized by the specific change:[31]
- Tautomerism — A base is changed by the repositioning of a hydrogen atom, altering the hydrogen bonding pattern of that base, resulting in incorrect base pairing during replication.
- Depurination — Loss of a purine base (A or G) to form an apurinic site (AP site).
- Deamination — Hydrolysis changes a normal base to an atypical base containing a keto group in place of the original amine group. Examples include C → U and A → HX (hypoxanthine), which can be corrected by DNA repair mechanisms; and 5MeC (5-methylcytosine) → T, which is less likely to be detected as a mutation because thymine is a normal DNA base.
- Slipped strand mispairing — Denaturation of the new strand from the template during replication, followed by renaturation in a different spot ("slipping"). This can lead to insertions or deletions.
- Replication slippage
Error-prone replication bypass
There is increasing evidence that the majority of spontaneously arising mutations are due to error-prone replication (translesion synthesis) past DNA damage in the template strand. Naturally occurring oxidative DNA damages arise at least 10,000 times per cell per day in humans and 50,000 times or more per cell per day in rats. In mice, the majority of mutations are caused by translesion synthesis.[32] Likewise, in yeast, Kunz et al.[33] found that more than 60% of the spontaneous single base pair substitutions and deletions were caused by translesion synthesis.
Errors introduced during DNA repair
Although naturally occurring double-strand breaks occur at a relatively low frequency in DNA, their repair often causes mutation. Non-homologous end joining (NHEJ) is a major pathway for repairing double-strand breaks. NHEJ involves removal of a few nucleotides to allow somewhat inaccurate alignment of the two ends for rejoining followed by addition of nucleotides to fill in gaps. As a consequence, NHEJ often introduces mutations.[34]
Induced mutation
Induced mutations are alterations in the gene after it has come in contact with mutagens and environmental causes.
Induced mutations on the molecular level can be caused by:
- Chemicals
- Hydroxylamine
- Base analogs (e.g., Bromodeoxyuridine (BrdU))
- Alkylating agents (e.g., N-ethyl-N-nitrosourea (ENU). These agents can mutate both replicating and non-replicating DNA. In contrast, a base analog can mutate the DNA only when the analog is incorporated in replicating the DNA. Each of these classes of chemical mutagens has certain effects that then lead to transitions, transversions, or deletions.
- Agents that form DNA adducts (e.g., ochratoxin A)[36]
- DNA intercalating agents (e.g., ethidium bromide)
- DNA crosslinkers
- Oxidative damage
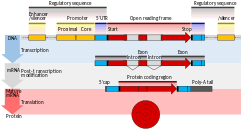 The transcriptional processes of splicing and translation of a eukaryotic gene
The transcriptional processes of splicing and translation of a eukaryotic gene - Nitrous acid converts amine groups on A and C to diazo groups, altering their hydrogen bonding patterns, which leads to incorrect base pairing during replication.
- Radiation
- Ultraviolet light (UV) (non-ionizing radiation). Two nucleotide bases in DNA—cytosine and thymine—are most vulnerable to radiation that can change their properties. UV light can induce adjacent pyrimidine bases in a DNA strand to become covalently joined as a pyrimidine dimer. UV radiation, in particular longer-wave UVA, can also cause oxidative damage to DNA.[37]
- Ionizing radiation. Exposure to ionizing radiation, such as gamma radiation, can result in mutation, possibly resulting in cancer or death.
Whereas in former times mutations were assumed to occur by chance, or induced by mutagens, molecular mechanisms of mutation have been discovered in bacteria and across the tree of life. As S. Rosenberg states, "These mechanisms reveal a picture of highly regulated mutagenesis, up-regulated temporally by stress responses and activated when cells/organisms are maladapted to their environments—when stressed—potentially accelerating adaptation." [38] Since they are self-induced mutagenic mechanisms that increase the adaptation rate of organisms, they have some times been named as adaptive mutagenesis mechanisms, and include the SOS response in bacteria,[39] ectopic intrachromosomal recombination [40] and other chromosomal events such as duplications.[38]
Classification of types
By effect on structure
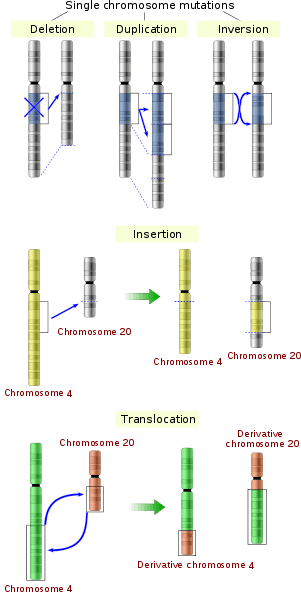

The sequence of a gene can be altered in a number of ways.[42] Gene mutations have varying effects on health depending on where they occur and whether they alter the function of essential proteins. Mutations in the structure of genes can be classified into several types.
Large-scale mutations
Large-scale mutations in chromosomal structure include:
- Amplifications (or gene duplications) or repetition of a chromosomal segment or presence of extra piece of a chromosome broken piece of a chromosome may become attached to a homologous or non-homologous chromosome so that some of the genes are present in more than two doses leading to multiple copies of all chromosomal regions, increasing the dosage of the genes located within them.
- Deletions of large chromosomal regions, leading to loss of the genes within those regions.
- Mutations whose effect is to juxtapose previously separate pieces of DNA, potentially bringing together separate genes to form functionally distinct fusion genes (e.g., bcr-abl).
- Large scale changes to the structure of chromosomes called chromosomal rearrangement that can lead to a decrease of fitness but also to speciation in isolated, inbred populations. These include:
- Chromosomal translocations: interchange of genetic parts from nonhomologous chromosomes.
- Chromosomal inversions: reversing the orientation of a chromosomal segment.
- Non-homologous chromosomal crossover.
- Interstitial deletions: an intra-chromosomal deletion that removes a segment of DNA from a single chromosome, thereby apposing previously distant genes. For example, cells isolated from a human astrocytoma, a type of brain tumor, were found to have a chromosomal deletion removing sequences between the Fused in Glioblastoma (FIG) gene and the receptor tyrosine kinase (ROS), producing a fusion protein (FIG-ROS). The abnormal FIG-ROS fusion protein has constitutively active kinase activity that causes oncogenic transformation (a transformation from normal cells to cancer cells).
- Loss of heterozygosity: loss of one allele, either by a deletion or a genetic recombination event, in an organism that previously had two different alleles.
Small-scale mutations
Small-scale mutations affect a gene in one or a few nucleotides. (If only a single nucleotide is affected, they are called point mutations.) Small-scale mutations include:
- Insertions add one or more extra nucleotides into the DNA. They are usually caused by transposable elements, or errors during replication of repeating elements. Insertions in the coding region of a gene may alter splicing of the mRNA (splice site mutation), or cause a shift in the reading frame (frameshift), both of which can significantly alter the gene product. Insertions can be reversed by excision of the transposable element.
- Deletions remove one or more nucleotides from the DNA. Like insertions, these mutations can alter the reading frame of the gene. In general, they are irreversible: Though exactly the same sequence might, in theory, be restored by an insertion, transposable elements able to revert a very short deletion (say 1–2 bases) in any location either are highly unlikely to exist or do not exist at all.
- Substitution mutations, often caused by chemicals or malfunction of DNA replication, exchange a single nucleotide for another.[43] These changes are classified as transitions or transversions.[44] Most common is the transition that exchanges a purine for a purine (A ↔ G) or a pyrimidine for a pyrimidine, (C ↔ T). A transition can be caused by nitrous acid, base mispairing, or mutagenic base analogs such as BrdU. Less common is a transversion, which exchanges a purine for a pyrimidine or a pyrimidine for a purine (C/T ↔ A/G). An example of a transversion is the conversion of adenine (A) into a cytosine (C). Point mutations are modifications of single base pairs of DNA or other small base pairs within a gene. A point mutation can be reversed by another point mutation, in which the nucleotide is changed back to its original state (true reversion) or by second-site reversion (a complementary mutation elsewhere that results in regained gene functionality). As discussed below, point mutations that occur within the protein coding region of a gene may be classified as synonymous or nonsynonymous substitutions, the latter of which in turn can be divided into missense or nonsense mutations.
By impact on protein sequence
- A frameshift mutation is a mutation caused by insertion or deletion of a number of nucleotides that is not evenly divisible by three from a DNA sequence. Due to the triplet nature of gene expression by codons, the insertion or deletion can disrupt the reading frame, or the grouping of the codons, resulting in a completely different translation from the original.[45] The earlier in the sequence the deletion or insertion occurs, the more altered the protein produced is. (For example, the code CCU GAC UAC CUA codes for the amino acids proline, aspartic acid, tyrosine, and leucine. If the U in CCU was deleted, the resulting sequence would be CCG ACU ACC UAx, which would instead code for proline, threonine, threonine, and part of another amino acid or perhaps a stop codon (where the x stands for the following nucleotide).) By contrast, any insertion or deletion that is evenly divisible by three is termed an in-frame mutation.
- A point substitution mutation results in a change in a single nucleotide and can be either synonymous or nonsynonymous.
- A synonymous substitution replaces a codon with another codon that codes for the same amino acid, so that the produced amino acid sequence is not modified. Synonymous mutations occur due to the degenerate nature of the genetic code. If this mutation does not result in any phenotypic effects, then it is called silent, but not all synonymous substitutions are silent. (There can also be silent mutations in nucleotides outside of the coding regions, such as the introns, because the exact nucleotide sequence is not as crucial as it is in the coding regions, but these are not considered synonymous substitutions.)
- A nonsynonymous substitution replaces a codon with another codon that codes for a different amino acid, so that the produced amino acid sequence is modified. Nonsynonymous substitutions can be classified as nonsense or missense mutations:
- A missense mutation changes a nucleotide to cause substitution of a different amino acid. This in turn can render the resulting protein nonfunctional. Such mutations are responsible for diseases such as Epidermolysis bullosa, sickle-cell disease, and SOD1-mediated ALS.[46] On the other hand, if a missense mutation occurs in an amino acid codon that results in the use of a different, but chemically similar, amino acid, then sometimes little or no change is rendered in the protein. For example, a change from AAA to AGA will encode arginine, a chemically similar molecule to the intended lysine. In this latter case the mutation will have little or no effect on phenotype and therefore be neutral.
- A nonsense mutation is a point mutation in a sequence of DNA that results in a premature stop codon, or a nonsense codon in the transcribed mRNA, and possibly a truncated, and often nonfunctional protein product. This sort of mutation has been linked to different mutations, such as congenital adrenal hyperplasia. (See Stop codon.)
By effect on function
- Loss-of-function mutations, also called inactivating mutations, result in the gene product having less or no function (being partially or wholly inactivated). When the allele has a complete loss of function (null allele), it is often called an amorph or amorphic mutation in the Muller's morphs schema. Phenotypes associated with such mutations are most often recessive. Exceptions are when the organism is haploid, or when the reduced dosage of a normal gene product is not enough for a normal phenotype (this is called haploinsufficiency).
- Gain-of-function mutations, also called activating mutations, change the gene product such that its effect gets stronger (enhanced activation) or even is superseded by a different and abnormal function. When the new allele is created, a heterozygote containing the newly created allele as well as the original will express the new allele; genetically this defines the mutations as dominant phenotypes. Several of Muller's morphs correspond to gain of function, including hypermorph (increased gene expression) and neomorph (novel function). In December 2017, the U.S. government lifted a temporary ban implemented in 2014 that banned federal funding for any new "gain-of-function" experiments that enhance pathogens "such as Avian influenza, SARS and the Middle East Respiratory Syndrome or MERS viruses."[47]
- Dominant negative mutations (also called antimorphic mutations) have an altered gene product that acts antagonistically to the wild-type allele. These mutations usually result in an altered molecular function (often inactive) and are characterized by a dominant or semi-dominant phenotype. In humans, dominant negative mutations have been implicated in cancer (e.g., mutations in genes p53,[48] ATM,[49] CEBPA[50] and PPARgamma[51]). Marfan syndrome is caused by mutations in the FBN1 gene, located on chromosome 15, which encodes fibrillin-1, a glycoprotein component of the extracellular matrix.[52] Marfan syndrome is also an example of dominant negative mutation and haploinsufficiency.[53][54]
- Hypomorphs, after Mullerian classification, are characterized by altered gene products that acts with decreased gene expression compared to the wild type allele. Usually, hypomorphic mutations are recessive, but haploinsufficiency causes some alleles to be dominant.
- Neomorphs are characterized by the control of new protein product synthesis.
- Lethal mutations are mutations that lead to the death of the organisms that carry the mutations.
- A back mutation or reversion is a point mutation that restores the original sequence and hence the original phenotype.[55]
By effect on fitness
In applied genetics, it is usual to speak of mutations as either harmful or beneficial.
- A harmful, or deleterious, mutation decreases the fitness of the organism.
- A beneficial, or advantageous mutation increases the fitness of the organism.
- A neutral mutation has no harmful or beneficial effect on the organism. Such mutations occur at a steady rate, forming the basis for the molecular clock. In the neutral theory of molecular evolution, neutral mutations provide genetic drift as the basis for most variation at the molecular level.
- A nearly neutral mutation is a mutation that may be slightly deleterious or advantageous, although most nearly neutral mutations are slightly deleterious.
Distribution of fitness effects
Attempts have been made to infer the distribution of fitness effects (DFE) using mutagenesis experiments and theoretical models applied to molecular sequence data. DFE, as used to determine the relative abundance of different types of mutations (i.e., strongly deleterious, nearly neutral or advantageous), is relevant to many evolutionary questions, such as the maintenance of genetic variation,[56] the rate of genomic decay,[57] the maintenance of outcrossing sexual reproduction as opposed to inbreeding[58] and the evolution of sex and genetic recombination.[59] DFE can also be tracked by tracking the skewness of the distribution of mutations with putatively severe effects as compared to the distribution of mutations with putatively mild or absent effect.[60] In summary, the DFE plays an important role in predicting evolutionary dynamics.[61][62] A variety of approaches have been used to study the DFE, including theoretical, experimental and analytical methods.
- Mutagenesis experiment: The direct method to investigate the DFE is to induce mutations and then measure the mutational fitness effects, which has already been done in viruses, bacteria, yeast, and Drosophila. For example, most studies of the DFE in viruses used site-directed mutagenesis to create point mutations and measure relative fitness of each mutant.[63][64][65][66] In Escherichia coli, one study used transposon mutagenesis to directly measure the fitness of a random insertion of a derivative of Tn10.[67] In yeast, a combined mutagenesis and deep sequencing approach has been developed to generate high-quality systematic mutant libraries and measure fitness in high throughput.[68] However, given that many mutations have effects too small to be detected[69] and that mutagenesis experiments can detect only mutations of moderately large effect; DNA sequence data analysis can provide valuable information about these mutations.
- Molecular sequence analysis: With rapid development of DNA sequencing technology, an enormous amount of DNA sequence data is available and even more is forthcoming in the future. Various methods have been developed to infer the DFE from DNA sequence data.[70][71][72][73] By examining DNA sequence differences within and between species, we are able to infer various characteristics of the DFE for neutral, deleterious and advantageous mutations.[24] To be specific, the DNA sequence analysis approach allows us to estimate the effects of mutations with very small effects, which are hardly detectable through mutagenesis experiments.
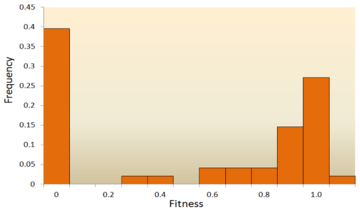
One of the earliest theoretical studies of the distribution of fitness effects was done by Motoo Kimura, an influential theoretical population geneticist. His neutral theory of molecular evolution proposes that most novel mutations will be highly deleterious, with a small fraction being neutral.[74][25] Hiroshi Akashi more recently proposed a bimodal model for the DFE, with modes centered around highly deleterious and neutral mutations.[75] Both theories agree that the vast majority of novel mutations are neutral or deleterious and that advantageous mutations are rare, which has been supported by experimental results. One example is a study done on the DFE of random mutations in vesicular stomatitis virus.[63] Out of all mutations, 39.6% were lethal, 31.2% were non-lethal deleterious, and 27.1% were neutral. Another example comes from a high throughput mutagenesis experiment with yeast.[68] In this experiment it was shown that the overall DFE is bimodal, with a cluster of neutral mutations, and a broad distribution of deleterious mutations.
Though relatively few mutations are advantageous, those that are play an important role in evolutionary changes.[76] Like neutral mutations, weakly selected advantageous mutations can be lost due to random genetic drift, but strongly selected advantageous mutations are more likely to be fixed. Knowing the DFE of advantageous mutations may lead to increased ability to predict the evolutionary dynamics. Theoretical work on the DFE for advantageous mutations has been done by John H. Gillespie[77] and H. Allen Orr.[78] They proposed that the distribution for advantageous mutations should be exponential under a wide range of conditions, which, in general, has been supported by experimental studies, at least for strongly selected advantageous mutations.[79][80][81]
In general, it is accepted that the majority of mutations are neutral or deleterious, with advantageous mutations being rare; however, the proportion of types of mutations varies between species. This indicates two important points: first, the proportion of effectively neutral mutations is likely to vary between species, resulting from dependence on effective population size; second, the average effect of deleterious mutations varies dramatically between species.[24] In addition, the DFE also differs between coding regions and noncoding regions, with the DFE of noncoding DNA containing more weakly selected mutations.[24]
By inheritance

In multicellular organisms with dedicated reproductive cells, mutations can be subdivided into germline mutations, which can be passed on to descendants through their reproductive cells, and somatic mutations (also called acquired mutations),[82] which involve cells outside the dedicated reproductive group and which are not usually transmitted to descendants.
Diploid organisms (e.g., humans) contain two copies of each gene—a paternal and a maternal allele. Based on the occurrence of mutation on each chromosome, we may classify mutations into three types. A wild type or homozygous non-mutated organism is one in which neither allele is mutated.
- A heterozygous mutation is a mutation of only one allele.
- A homozygous mutation is an identical mutation of both the paternal and maternal alleles.
- Compound heterozygous mutations or a genetic compound consists of two different mutations in the paternal and maternal alleles.[83]
Germline mutation
A germline mutation in the reproductive cells of an individual gives rise to a constitutional mutation in the offspring, that is, a mutation that is present in every cell. A constitutional mutation can also occur very soon after fertilisation, or continue from a previous constitutional mutation in a parent.[84] A germline mutation can be passed down through subsequent generations of organisms.
The distinction between germline and somatic mutations is important in animals that have a dedicated germline to produce reproductive cells. However, it is of little value in understanding the effects of mutations in plants, which lack a dedicated germline. The distinction is also blurred in those animals that reproduce asexually through mechanisms such as budding, because the cells that give rise to the daughter organisms also give rise to that organism's germline.
A new germline mutation not inherited from either parent is called a de novo mutation.
Somatic mutation
A change in the genetic structure that is not inherited from a parent, and also not passed to offspring, is called a somatic mutation.[82] Somatic mutations are not inherited by an organism's offspring because they do not affect the germline. However, they are passed down to the all the progeny of a mutated cell within the same organism during mitosis. A major section of an organism therefore might carry the same mutation. These types of mutations are usually prompted by environmental causes, such as ultraviolet radiation or any exposure to certain harmful chemicals, and can cause diseases including cancer.[85]
With plants, some somatic mutations can be propagated without the need for seed production, for example, by grafting and stem cuttings. These type of mutation have led to new types of fruits, such as the "Delicious" apple and the "Washington" navel orange.[86]
Human and mouse somatic cells have a mutation rate more than ten times higher than the germline mutation rate for both species; mice have a higher rate of both somatic and germline mutations per cell division than humans. The disparity in mutation rate between the germline and somatic tissues likely reflects the greater importance of genome maintenance in the germline than in the soma.[87]
Special classes
- Conditional mutation is a mutation that has wild-type (or less severe) phenotype under certain "permissive" environmental conditions and a mutant phenotype under certain "restrictive" conditions. For example, a temperature-sensitive mutation can cause cell death at high temperature (restrictive condition), but might have no deleterious consequences at a lower temperature (permissive condition).[88] These mutations are non-autonomous, as their manifestation depends upon presence of certain conditions, as opposed to other mutations which appear autonomously.[89] The permissive conditions may be temperature,[90] certain chemicals,[91] light[91] or mutations in other parts of the genome.[89] In vivo mechanisms like transcriptional switches can create conditional mutations. For instance, association of Steroid Binding Domain can create a transcriptional switch that can change the expression of a gene based on the presence of a steroid ligand.[92] Conditional mutations have applications in research as they allow control over gene expression. This is especially useful studying diseases in adults by allowing expression after a certain period of growth, thus eliminating the deleterious effect of gene expression seen during stages of development in model organisms.[91] DNA Recombinase systems like Cre-Lox Recombination used in association with promoters that are activated under certain conditions can generate conditional mutations. Dual Recombinase technology can be used to induce multiple conditional mutations to study the diseases which manifest as a result of simultaneous mutations in multiple genes.[91] Certain inteins have been identified which splice only at certain permissive temperatures, leading to improper protein synthesis and thus, loss-of-function mutations at other temperatures.[93] Conditional mutations may also be used in genetic studies associated with ageing, as the expression can be changed after a certain time period in the organism's lifespan.[90]
- Replication timing quantitative trait loci affects DNA replication.
Nomenclature
In order to categorize a mutation as such, the "normal" sequence must be obtained from the DNA of a "normal" or "healthy" organism (as opposed to a "mutant" or "sick" one), it should be identified and reported; ideally, it should be made publicly available for a straightforward nucleotide-by-nucleotide comparison, and agreed upon by the scientific community or by a group of expert geneticists and biologists, who have the responsibility of establishing the standard or so-called "consensus" sequence. This step requires a tremendous scientific effort. Once the consensus sequence is known, the mutations in a genome can be pinpointed, described, and classified. The committee of the Human Genome Variation Society (HGVS) has developed the standard human sequence variant nomenclature,[94] which should be used by researchers and DNA diagnostic centers to generate unambiguous mutation descriptions. In principle, this nomenclature can also be used to describe mutations in other organisms. The nomenclature specifies the type of mutation and base or amino acid changes.
- Nucleotide substitution (e.g., 76A>T) — The number is the position of the nucleotide from the 5' end; the first letter represents the wild-type nucleotide, and the second letter represents the nucleotide that replaced the wild type. In the given example, the adenine at the 76th position was replaced by a thymine.
- If it becomes necessary to differentiate between mutations in genomic DNA, mitochondrial DNA, and RNA, a simple convention is used. For example, if the 100th base of a nucleotide sequence mutated from G to C, then it would be written as g.100G>C if the mutation occurred in genomic DNA, m.100G>C if the mutation occurred in mitochondrial DNA, or r.100g>c if the mutation occurred in RNA. Note that, for mutations in RNA, the nucleotide code is written in lower case.
- Amino acid substitution (e.g., D111E) — The first letter is the one letter code of the wild-type amino acid, the number is the position of the amino acid from the N-terminus, and the second letter is the one letter code of the amino acid present in the mutation. Nonsense mutations are represented with an X for the second amino acid (e.g. D111X).
- Amino acid deletion (e.g., ΔF508) — The Greek letter Δ (delta) indicates a deletion. The letter refers to the amino acid present in the wild type and the number is the position from the N terminus of the amino acid were it to be present as in the wild type.
Mutation rates
Mutation rates vary substantially across species, and the evolutionary forces that generally determine mutation are the subject of ongoing investigation.
The genomes of RNA viruses are based on RNA rather than DNA. The RNA viral genome can be double-stranded (as in DNA) or single-stranded. In some of these viruses (such as the single-stranded human immunodeficiency virus), replication occurs quickly, and there are no mechanisms to check the genome for accuracy. This error-prone process often results in mutations.
Disease causation
Changes in DNA caused by mutation in a coding region of DNA can cause errors in protein sequence that may result in partially or completely non-functional proteins. Each cell, in order to function correctly, depends on thousands of proteins to function in the right places at the right times. When a mutation alters a protein that plays a critical role in the body, a medical condition can result. Some mutations alter a gene's DNA base sequence but do not change the function of the protein made by the gene. One study on the comparison of genes between different species of Drosophila suggests that if a mutation does change a protein, the mutation will most likely be harmful, with an estimated 70 percent of amino acid polymorphisms having damaging effects, and the remainder being either neutral or weakly beneficial.[8] However, studies have shown that only 7% of point mutations in noncoding DNA of yeast are deleterious and 12% in coding DNA are deleterious. The rest of the mutations are either neutral or slightly beneficial.[95]
Inherited disorders
If a mutation is present in a germ cell, it can give rise to offspring that carries the mutation in all of its cells. This is the case in hereditary diseases. In particular, if there is a mutation in a DNA repair gene within a germ cell, humans carrying such germline mutations may have an increased risk of cancer. A list of 34 such germline mutations is given in the article DNA repair-deficiency disorder. An example of one is albinism, a mutation that occurs in the OCA1 or OCA2 gene. Individuals with this disorder are more prone to many types of cancers, other disorders and have impaired vision.
DNA damage can cause an error when the DNA is replicated, and this error of replication can cause a gene mutation that, in turn, could cause a genetic disorder. DNA damages are repaired by the DNA repair system of the cell. Each cell has a number of pathways through which enzymes recognize and repair damages in DNA. Because DNA can be damaged in many ways, the process of DNA repair is an important way in which the body protects itself from disease. Once DNA damage has given rise to a mutation, the mutation cannot be repaired.
Role in carcinogenesis
On the other hand, a mutation may occur in a somatic cell of an organism. Such mutations will be present in all descendants of this cell within the same organism. The accumulation of certain mutations over generations of somatic cells is part of cause of malignant transformation, from normal cell to cancer cell.[96]
Cells with heterozygous loss-of-function mutations (one good copy of gene and one mutated copy) may function normally with the unmutated copy until the good copy has been spontaneously somatically mutated. This kind of mutation happens often in living organisms, but it is difficult to measure the rate. Measuring this rate is important in predicting the rate at which people may develop cancer.[97]
Point mutations may arise from spontaneous mutations that occur during DNA replication. The rate of mutation may be increased by mutagens. Mutagens can be physical, such as radiation from UV rays, X-rays or extreme heat, or chemical (molecules that misplace base pairs or disrupt the helical shape of DNA). Mutagens associated with cancers are often studied to learn about cancer and its prevention.
Prion mutations
Prions are proteins and do not contain genetic material. However, prion replication has been shown to be subject to mutation and natural selection just like other forms of replication.[98] The human gene PRNP codes for the major prion protein, PrP, and is subject to mutations that can give rise to disease-causing prions.
Beneficial mutations
Although mutations that cause changes in protein sequences can be harmful to an organism, on occasions the effect may be positive in a given environment. In this case, the mutation may enable the mutant organism to withstand particular environmental stresses better than wild-type organisms, or reproduce more quickly. In these cases a mutation will tend to become more common in a population through natural selection. Examples include the following:
HIV resistance: a specific 32 base pair deletion in human CCR5 (CCR5-Δ32) confers HIV resistance to homozygotes and delays AIDS onset in heterozygotes.[99] One possible explanation of the etiology of the relatively high frequency of CCR5-Δ32 in the European population is that it conferred resistance to the bubonic plague in mid-14th century Europe. People with this mutation were more likely to survive infection; thus its frequency in the population increased.[100] This theory could explain why this mutation is not found in Southern Africa, which remained untouched by bubonic plague. A newer theory suggests that the selective pressure on the CCR5 Delta 32 mutation was caused by smallpox instead of the bubonic plague.[101]
Malaria resistance: An example of a harmful mutation is sickle-cell disease, a blood disorder in which the body produces an abnormal type of the oxygen-carrying substance hemoglobin in the red blood cells. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the allele, because, in areas where malaria is common, there is a survival value in carrying only a single sickle-cell allele (sickle cell trait).[102] Those with only one of the two alleles of the sickle-cell disease are more resistant to malaria, since the infestation of the malaria Plasmodium is halted by the sickling of the cells that it infests.
Antibiotic resistance: Practically all bacteria develop antibiotic resistance when exposed to antibiotics. In fact, bacterial populations already have such mutations that get selected under antibiotic selection.[103] Obviously, such mutations are only beneficial for the bacteria but not for those infected.
Lactase persistence. A mutation allowed humans to express the enzyme lactase after they are naturally weaned from breast milk, allowing adults to digest lactose, which is likely one of the most beneficial mutations in recent human evolution.[104]
History
%2C_by_Th%C3%A9r%C3%A8se_Schwartze_(1851-1918).jpg)
Mutationism is one of several alternatives to evolution by natural selection that have existed both before and after the publication of Charles Darwin's 1859 book, On the Origin of Species. In the theory, mutation was the source of novelty, creating new forms and new species, potentially instantaneously,[105] in a sudden jump.[106] This was envisaged as driving evolution, which was limited by the supply of mutations.
Before Darwin, biologists commonly believed in saltationism, the possibility of large evolutionary jumps, including immediate speciation. For example, in 1822 Étienne Geoffroy Saint-Hilaire argued that species could be formed by sudden transformations, or what would later be called macromutation.[107] Darwin opposed saltation, insisting on gradualism in evolution as in geology. In 1864, Albert von Kölliker revived Geoffroy's theory.[108] In 1901 the geneticist Hugo de Vries gave the name "mutation" to seemingly new forms that suddenly arose in his experiments on the evening primrose Oenothera lamarckiana, and in the first decade of the 20th century, mutationism, or as de Vries named it mutationstheorie,[109][105] became a rival to Darwinism supported for a while by geneticists including William Bateson,[110] Thomas Hunt Morgan, and Reginald Punnett.[111][105]
Understanding of mutationism is clouded by the mid-20th century portrayal of the early mutationists by supporters of the modern synthesis as opponents of Darwinian evolution and rivals of the biometrics school who argued that selection operated on continuous variation. In this portrayal, mutationism was defeated by a synthesis of genetics and natural selection that supposedly started later, around 1918, with work by the mathematician Ronald Fisher.[112][113][114][115] However, the alignment of Mendelian genetics and natural selection began as early as 1902 with a paper by Udny Yule,[116] and built up with theoretical and experimental work in Europe and America. Despite the controversy, the early mutationists had by 1918 already accepted natural selection and explained continuous variation as the result of multiple genes acting on the same characteristic, such as height.[113][114]
Mutationism, along with other alternatives to Darwinism like Lamarckism and orthogenesis, was discarded by most biologists as they came to see that Mendelian genetics and natural selection could readily work together; mutation took its place as a source of the genetic variation essential for natural selection to work on. However, mutationism did not entirely vanish. In 1940, Richard Goldschmidt again argued for single-step speciation by macromutation, describing the organisms thus produced as "hopeful monsters", earning widespread ridicule.[117][118] In 1987, Masatoshi Nei argued controversially that evolution was often mutation-limited.[119] Modern biologists such as Douglas J. Futuyma conclude that essentially all claims of evolution driven by large mutations can be explained by Darwinian evolution.[120]
See also
- Aneuploidy
- Antioxidant
- Budgerigar colour genetics
- Deletion (genetics)
- Ecogenetics
- Embryology
- Homeobox
- Polyploidy
- Robertsonian translocation
- Signature-tagged mutagenesis
- Somatic hypermutation
- TILLING (molecular biology)
- Trinucleotide repeat expansion
- Human somatic variations
References
- "mutation | Learn Science at Scitable". www.nature.com. Retrieved 24 September 2018.
- Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC (March 2015). "Homology and enzymatic requirements of microhomology-dependent alternative end joining". Cell Death & Disease. 6 (3): e1697. doi:10.1038/cddis.2015.58. PMC 4385936. PMID 25789972.
- Chen J, Miller BF, Furano AV (April 2014). "Repair of naturally occurring mismatches can induce mutations in flanking DNA". eLife. 3: e02001. doi:10.7554/elife.02001. PMC 3999860. PMID 24843013.
- Rodgers K, McVey M (January 2016). "Error-Prone Repair of DNA Double-Strand Breaks". Journal of Cellular Physiology. 231 (1): 15–24. doi:10.1002/jcp.25053. PMC 4586358. PMID 26033759.
- Bertram JS (December 2000). "The molecular biology of cancer". Molecular Aspects of Medicine. 21 (6): 167–223. doi:10.1016/S0098-2997(00)00007-8. PMID 11173079.
- Aminetzach YT, Macpherson JM, Petrov DA (July 2005). "Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila". Science. 309 (5735): 764–7. Bibcode:2005Sci...309..764A. doi:10.1126/science.1112699. PMID 16051794.
- Burrus V, Waldor MK (June 2004). "Shaping bacterial genomes with integrative and conjugative elements". Research in Microbiology. 155 (5): 376–86. doi:10.1016/j.resmic.2004.01.012. PMID 15207870.
- Sawyer SA, Parsch J, Zhang Z, Hartl DL (April 2007). "Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila". Proceedings of the National Academy of Sciences of the United States of America. 104 (16): 6504–10. Bibcode:2007PNAS..104.6504S. doi:10.1073/pnas.0701572104. PMC 1871816. PMID 17409186.
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G (August 2009). "Mechanisms of change in gene copy number". Nature Reviews. Genetics. 10 (8): 551–64. doi:10.1038/nrg2593. PMC 2864001. PMID 19597530.
- Carroll SB, Grenier JK, Weatherbee SD (2005). From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design (2nd ed.). Malden, MA: Blackwell Publishing. ISBN 978-1-4051-1950-4. LCCN 2003027991. OCLC 53972564.
- Harrison PM, Gerstein M (May 2002). "Studying genomes through the aeons: protein families, pseudogenes and proteome evolution". Journal of Molecular Biology. 318 (5): 1155–74. doi:10.1016/S0022-2836(02)00109-2. PMID 12083509.
- Orengo CA, Thornton JM (July 2005). "Protein families and their evolution-a structural perspective". Annual Review of Biochemistry. 74: 867–900. doi:10.1146/annurev.biochem.74.082803.133029. PMID 15954844.
- Long M, Betrán E, Thornton K, Wang W (November 2003). "The origin of new genes: glimpses from the young and old". Nature Reviews. Genetics. 4 (11): 865–75. doi:10.1038/nrg1204. PMID 14634634.
- Wang M, Caetano-Anollés G (January 2009). "The evolutionary mechanics of domain organization in proteomes and the rise of modularity in the protein world". Structure. 17 (1): 66–78. doi:10.1016/j.str.2008.11.008. PMID 19141283.
- Bowmaker JK (May 1998). "Evolution of colour vision in vertebrates". Eye. 12 (Pt 3b): 541–7. doi:10.1038/eye.1998.143. PMID 9775215.
- Gregory TR, Hebert PD (April 1999). "The modulation of DNA content: proximate causes and ultimate consequences". Genome Research. 9 (4): 317–24. doi:10.1101/gr.9.4.317 (inactive 24 March 2020). PMID 10207154.
- Hurles M (July 2004). "Gene duplication: the genomic trade in spare parts". PLOS Biology. 2 (7): E206. doi:10.1371/journal.pbio.0020206. PMC 449868. PMID 15252449.
- Liu N, Okamura K, Tyler DM, Phillips MD, Chung WJ, Lai EC (October 2008). "The evolution and functional diversification of animal microRNA genes". Cell Research. 18 (10): 985–96. doi:10.1038/cr.2008.278. PMC 2712117. PMID 18711447.
- Siepel A (October 2009). "Darwinian alchemy: Human genes from noncoding DNA". Genome Research. 19 (10): 1693–5. doi:10.1101/gr.098376.109. PMC 2765273. PMID 19797681.
- Zhang J, Wang X, Podlaha O (May 2004). "Testing the chromosomal speciation hypothesis for humans and chimpanzees". Genome Research. 14 (5): 845–51. doi:10.1101/gr.1891104. PMC 479111. PMID 15123584.
- Ayala FJ, Coluzzi M (May 2005). "Chromosome speciation: humans, Drosophila, and mosquitoes". Proceedings of the National Academy of Sciences of the United States of America. 102 Suppl 1 (Suppl 1): 6535–42. Bibcode:2005PNAS..102.6535A. doi:10.1073/pnas.0501847102. PMC 1131864. PMID 15851677.
- Hurst GD, Werren JH (August 2001). "The role of selfish genetic elements in eukaryotic evolution". Nature Reviews Genetics. 2 (8): 597–606. doi:10.1038/35084545. PMID 11483984.
- Häsler J, Strub K (November 2006). "Alu elements as regulators of gene expression". Nucleic Acids Research. 34 (19): 5491–7. doi:10.1093/nar/gkl706. PMC 1636486. PMID 17020921.
- Eyre-Walker A, Keightley PD (August 2007). "The distribution of fitness effects of new mutations" (PDF). Nature Reviews Genetics. 8 (8): 610–8. doi:10.1038/nrg2146. PMID 17637733. Archived (PDF) from the original on 4 March 2016.
- Kimura M (1983). The Neutral Theory of Molecular Evolution. Cambridge, UK; New York: Cambridge University Press. ISBN 978-0-521-23109-1. LCCN 82022225. OCLC 9081989.CS1 maint: ref=harv (link)
- Bohidar HB (January 2015). Fundamentals of Polymer Physics and Molecular Biophysics. Cambridge University Press. ISBN 978-1-316-09302-3.
- Dover GA, Darwin C (2000). Dear Mr. Darwin: Letters on the Evolution of Life and Human Nature. University of California Press. ISBN 9780520227903.
- Tibayrenc, Michel (12 January 2017). Genetics and Evolution of Infectious Diseases. Elsevier. ISBN 9780128001530.
- "Cancer Is Partly Caused By Bad Luck, Study Finds". Archived from the original on 13 July 2017.
- Jha, Alok (22 August 2012). "Older fathers pass on more genetic mutations, study shows". the Guardian.
- Montelone BA (1998). "Mutation, Mutagens, and DNA Repair". www-personal.ksu.edu. Archived from the original on 26 September 2015. Retrieved 2 October 2015.
- Stuart GR, Oda Y, de Boer JG, Glickman BW (March 2000). "Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice". Genetics. 154 (3): 1291–300. PMC 1460990. PMID 10757770.
- Kunz BA, Ramachandran K, Vonarx EJ (April 1998). "DNA sequence analysis of spontaneous mutagenesis in Saccharomyces cerevisiae". Genetics. 148 (4): 1491–505. PMC 1460101. PMID 9560369.
- Lieber MR (July 2010). "The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway". Annual Review of Biochemistry. 79: 181–211. doi:10.1146/annurev.biochem.052308.093131. PMC 3079308. PMID 20192759.
- Created from PDB 1JDG Archived 31 December 2015 at the Wayback Machine
- Pfohl-Leszkowicz A, Manderville RA (January 2007). "Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans". Molecular Nutrition & Food Research. 51 (1): 61–99. doi:10.1002/mnfr.200600137. PMID 17195275.
- Kozmin S, Slezak G, Reynaud-Angelin A, Elie C, de Rycke Y, Boiteux S, Sage E (September 2005). "UVA radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae". Proceedings of the National Academy of Sciences of the United States of America. 102 (38): 13538–43. Bibcode:2005PNAS..10213538K. doi:10.1073/pnas.0504497102. PMC 1224634. PMID 16157879.
- Fitzgerald DM, Rosenberg SM (April 2019). "What is mutation? A chapter in the series: How microbes "jeopardize" the modern synthesis". PLOS Genetics. 15 (4): e1007995. doi:10.1371/journal.pgen.1007995. PMC 6443146. PMID 30933985.
- Galhardo RS, Hastings PJ, Rosenberg SM (1 January 2007). "Mutation as a stress response and the regulation of evolvability". Critical Reviews in Biochemistry and Molecular Biology. 42 (5): 399–435. doi:10.1080/10409230701648502. PMC 3319127. PMID 17917874.
- Quinto-Alemany D, Canerina-Amaro A, Hernández-Abad LG, Machín F, Romesberg FE, Gil-Lamaignere C (31 July 2012). Sturtevant J (ed.). "Yeasts acquire resistance secondary to antifungal drug treatment by adaptive mutagenesis". PLOS One. 7 (7): e42279. Bibcode:2012PLoSO...742279Q. doi:10.1371/journal.pone.0042279. PMC 3409178. PMID 22860105.
- References for the image are found in Wikimedia Commons page at: Commons:File:Notable mutations.svg#References.
- Rahman, Nazneen. "The clinical impact of DNA sequence changes". Transforming Genetic Medicine Initiative. Archived from the original on 4 August 2017. Retrieved 27 June 2017.
- Freese E (April 1959). "The Difference Between Spontaneous and Base-Analogue Induced Mutations of Phage T4". Proceedings of the National Academy of Sciences of the United States of America. 45 (4): 622–33. Bibcode:1959PNAS...45..622F. doi:10.1073/pnas.45.4.622. PMC 222607. PMID 16590424.
- Freese E (June 1959). "The specific mutagenic effect of base analogues on Phage T4". Journal of Molecular Biology. 1 (2): 87–105. doi:10.1016/S0022-2836(59)80038-3.
- Hogan, C. Michael (12 October 2010). "Mutation". In Monosson, Emily (ed.). Encyclopedia of Earth. Washington, D.C.: Environmental Information Coalition, National Council for Science and the Environment. OCLC 72808636. Archived from the original on 14 November 2015. Retrieved 8 October 2015.
- Boillée S, Vande Velde C, Cleveland DW (October 2006). "ALS: a disease of motor neurons and their nonneuronal neighbors". Neuron. 52 (1): 39–59. CiteSeerX 10.1.1.325.7514. doi:10.1016/j.neuron.2006.09.018. PMID 17015226.
- Steenhuysen, Julie (19 December 2017). "U.S. Lifts Funding Ban on Studies That Enhance Dangerous Germs". U.S. News & World Report. Retrieved 15 January 2018.
- Goh AM, Coffill CR, Lane DP (January 2011). "The role of mutant p53 in human cancer". The Journal of Pathology. 223 (2): 116–26. doi:10.1002/path.2784. PMID 21125670.
- Chenevix-Trench G, Spurdle AB, Gatei M, Kelly H, Marsh A, Chen X, Donn K, Cummings M, Nyholt D, Jenkins MA, Scott C, Pupo GM, Dörk T, Bendix R, Kirk J, Tucker K, McCredie MR, Hopper JL, Sambrook J, Mann GJ, Khanna KK (February 2002). "Dominant negative ATM mutations in breast cancer families". Journal of the National Cancer Institute. 94 (3): 205–15. CiteSeerX 10.1.1.557.6394. doi:10.1093/jnci/94.3.205. PMID 11830610.
- Paz-Priel I, Friedman A (2011). "C/EBPα dysregulation in AML and ALL". Critical Reviews in Oncogenesis. 16 (1–2): 93–102. doi:10.1615/critrevoncog.v16.i1-2.90. PMC 3243939. PMID 22150310.
- Capaccio D, Ciccodicola A, Sabatino L, Casamassimi A, Pancione M, Fucci A, Febbraro A, Merlino A, Graziano G, Colantuoni V (June 2010). "A novel germline mutation in peroxisome proliferator-activated receptor gamma gene associated with large intestine polyp formation and dyslipidemia". Biochimica et Biophysica Acta. 1802 (6): 572–81. doi:10.1016/j.bbadis.2010.01.012. PMID 20123124.
- McKusick VA (July 1991). "The defect in Marfan syndrome". Nature. 352 (6333): 279–81. Bibcode:1991Natur.352..279M. doi:10.1038/352279a0. PMID 1852198.
- Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC (July 2004). "Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome". The Journal of Clinical Investigation. 114 (2): 172–81. doi:10.1172/JCI20641. PMC 449744. PMID 15254584.
- Judge DP, Dietz HC (December 2005). "Marfan's syndrome". Lancet. 366 (9501): 1965–76. doi:10.1016/S0140-6736(05)67789-6. PMC 1513064. PMID 16325700.
- Ellis NA, Ciocci S, German J (February 2001). "Back mutation can produce phenotype reversion in Bloom syndrome somatic cells". Human Genetics. 108 (2): 167–73. doi:10.1007/s004390000447. PMID 11281456.
- Charlesworth D, Charlesworth B, Morgan MT (December 1995). "The pattern of neutral molecular variation under the background selection model". Genetics. 141 (4): 1619–32. PMC 1206892. PMID 8601499.
- Loewe L (April 2006). "Quantifying the genomic decay paradox due to Muller's ratchet in human mitochondrial DNA". Genetical Research. 87 (2): 133–59. doi:10.1017/S0016672306008123. PMID 16709275.
- Bernstein H, Hopf FA, Michod RE (1987). The molecular basis of the evolution of sex. Advances in Genetics. 24. pp. 323–70. doi:10.1016/s0065-2660(08)60012-7. ISBN 9780120176243. PMID 3324702.
- Peck JR, Barreau G, Heath SC (April 1997). "Imperfect genes, Fisherian mutation and the evolution of sex". Genetics. 145 (4): 1171–99. PMC 1207886. PMID 9093868.
- Simcikova D, Heneberg P (December 2019). "Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases". Scientific Reports. 9 (1): 18577. Bibcode:2019NatSR...918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466. PMID 31819097.CS1 maint: uses authors parameter (link)
- Keightley PD, Lynch M (March 2003). "Toward a realistic model of mutations affecting fitness". Evolution; International Journal of Organic Evolution. 57 (3): 683–5, discussion 686–9. doi:10.1554/0014-3820(2003)057[0683:tarmom]2.0.co;2. JSTOR 3094781. PMID 12703958.
- Barton NH, Keightley PD (January 2002). "Understanding quantitative genetic variation". Nature Reviews Genetics. 3 (1): 11–21. doi:10.1038/nrg700. PMID 11823787.
- Sanjuán R, Moya A, Elena SF (June 2004). "The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus". Proceedings of the National Academy of Sciences of the United States of America. 101 (22): 8396–401. Bibcode:2004PNAS..101.8396S. doi:10.1073/pnas.0400146101. PMC 420405. PMID 15159545.
- Carrasco P, de la Iglesia F, Elena SF (December 2007). "Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus". Journal of Virology. 81 (23): 12979–84. doi:10.1128/JVI.00524-07. PMC 2169111. PMID 17898073.
- Sanjuán R (June 2010). "Mutational fitness effects in RNA and single-stranded DNA viruses: common patterns revealed by site-directed mutagenesis studies". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 365 (1548): 1975–82. doi:10.1098/rstb.2010.0063. PMC 2880115. PMID 20478892.
- Peris JB, Davis P, Cuevas JM, Nebot MR, Sanjuán R (June 2010). "Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1". Genetics. 185 (2): 603–9. doi:10.1534/genetics.110.115162. PMC 2881140. PMID 20382832.
- Elena SF, Ekunwe L, Hajela N, Oden SA, Lenski RE (March 1998). "Distribution of fitness effects caused by random insertion mutations in Escherichia coli". Genetica. 102–103 (1–6): 349–58. doi:10.1023/A:1017031008316. PMID 9720287.
- Hietpas RT, Jensen JD, Bolon DN (May 2011). "Experimental illumination of a fitness landscape". Proceedings of the National Academy of Sciences of the United States of America. 108 (19): 7896–901. Bibcode:2011PNAS..108.7896H. doi:10.1073/pnas.1016024108. PMC 3093508. PMID 21464309.
- Davies EK, Peters AD, Keightley PD (September 1999). "High frequency of cryptic deleterious mutations in Caenorhabditis elegans". Science. 285 (5434): 1748–51. doi:10.1126/science.285.5434.1748. PMID 10481013.
- Loewe L, Charlesworth B (September 2006). "Inferring the distribution of mutational effects on fitness in Drosophila". Biology Letters. 2 (3): 426–30. doi:10.1098/rsbl.2006.0481. PMC 1686194. PMID 17148422.
- Eyre-Walker A, Woolfit M, Phelps T (June 2006). "The distribution of fitness effects of new deleterious amino acid mutations in humans". Genetics. 173 (2): 891–900. doi:10.1534/genetics.106.057570. PMC 1526495. PMID 16547091.
- Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL (August 2003). "Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection". Journal of Molecular Evolution. 57 Suppl 1 (1): S154–64. Bibcode:2003JMolE..57S.154S. CiteSeerX 10.1.1.78.65. doi:10.1007/s00239-003-0022-3. PMID 15008412.
- Piganeau G, Eyre-Walker A (September 2003). "Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock". Proceedings of the National Academy of Sciences of the United States of America. 100 (18): 10335–40. Bibcode:2003PNAS..10010335P. doi:10.1073/pnas.1833064100. PMC 193562. PMID 12925735.
- Kimura M (February 1968). "Evolutionary rate at the molecular level". Nature. 217 (5129): 624–6. Bibcode:1968Natur.217..624K. doi:10.1038/217624a0. PMID 5637732.
- Akashi H (September 1999). "Within- and between-species DNA sequence variation and the 'footprint' of natural selection". Gene. 238 (1): 39–51. doi:10.1016/S0378-1119(99)00294-2. PMID 10570982.
- Eyre-Walker A (October 2006). "The genomic rate of adaptive evolution". Trends in Ecology & Evolution. 21 (10): 569–75. doi:10.1016/j.tree.2006.06.015. PMID 16820244.
- Gillespie JH (September 1984). "Molecular Evolution Over the Mutational Landscape". Evolution. 38 (5): 1116–1129. doi:10.2307/2408444. JSTOR 2408444. PMID 28555784.
- Orr HA (April 2003). "The distribution of fitness effects among beneficial mutations". Genetics. 163 (4): 1519–26. PMC 1462510. PMID 12702694.
- Kassen R, Bataillon T (April 2006). "Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria". Nature Genetics. 38 (4): 484–8. doi:10.1038/ng1751. PMID 16550173.
- Rokyta DR, Joyce P, Caudle SB, Wichman HA (April 2005). "An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus". Nature Genetics. 37 (4): 441–4. doi:10.1038/ng1535. PMID 15778707.
- Imhof M, Schlotterer C (January 2001). "Fitness effects of advantageous mutations in evolving Escherichia coli populations". Proceedings of the National Academy of Sciences of the United States of America. 98 (3): 1113–7. Bibcode:2001PNAS...98.1113I. doi:10.1073/pnas.98.3.1113. PMC 14717. PMID 11158603.
- "Somatic cell genetic mutation". Genome Dictionary. Athens, Greece: Information Technology Associates. 30 June 2007. Archived from the original on 24 February 2010. Retrieved 6 June 2010.
- "Compound heterozygote". MedTerms. New York: WebMD. 14 June 2012. Archived from the original on 4 March 2016. Retrieved 9 October 2015.
- "RB1 Genetics". Daisy's Eye Cancer Fund. Oxford, UK. Archived from the original on 26 November 2011. Retrieved 9 October 2015.
- "somatic mutation | genetics". Encyclopædia Britannica. Archived from the original on 31 March 2017. Retrieved 31 March 2017.
- Hartl, Jones, Daniel L., Elizabeth W. (1998). Genetics Principles and Analysis. Sudbury, Massachusetts: Jones and Bartlett Publishers. pp. 556. ISBN 978-0-7637-0489-6.
- Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J (2017). "Differences between germline and somatic mutation rates in humans and mice". Nat Commun. 8: 15183. Bibcode:2017NatCo...815183M. doi:10.1038/ncomms15183. PMC 5436103. PMID 28485371.
- Alberts (2014). Molecular Biology of the Cell (6 ed.). Garland Science. p. 487. ISBN 9780815344322.
- Chadov BF, Fedorova NB, Chadova EV (1 July 2015). "Conditional mutations in Drosophila melanogaster: On the occasion of the 150th anniversary of G. Mendel's report in Brünn". Mutation Research/Reviews in Mutation Research. 765: 40–55. doi:10.1016/j.mrrev.2015.06.001. PMID 26281767.
- Landis G, Bhole D, Lu L, Tower J (July 2001). "High-frequency generation of conditional mutations affecting Drosophila melanogaster development and life span". Genetics. 158 (3): 1167–76. PMC 1461716. PMID 11454765. Archived from the original on 22 March 2017.
- Gierut JJ, Jacks TE, Haigis KM (April 2014). "Strategies to achieve conditional gene mutation in mice". Cold Spring Harbor Protocols. 2014 (4): 339–49. doi:10.1101/pdb.top069807. PMC 4142476. PMID 24692485.
- Spencer DM (May 1996). "Creating conditional mutations in mammals". Trends in Genetics. 12 (5): 181–7. doi:10.1016/0168-9525(96)10013-5. PMID 8984733.
- Tan G, Chen M, Foote C, Tan C (September 2009). "Temperature-sensitive mutations made easy: generating conditional mutations by using temperature-sensitive inteins that function within different temperature ranges". Genetics. 183 (1): 13–22. doi:10.1534/genetics.109.104794. PMC 2746138. PMID 19596904.
- den Dunnen JT, Antonarakis SE (January 2000). "Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion". Human Mutation. 15 (1): 7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. PMID 10612815.
- Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, Yang SP, Fay JC (August 2008). Pritchard JK (ed.). "A catalog of neutral and deleterious polymorphism in yeast". PLOS Genetics. 4 (8): e1000183. doi:10.1371/journal.pgen.1000183. PMC 2515631. PMID 18769710.
- Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M (June 1993). "Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis". Nature. 363 (6429): 558–61. Bibcode:1993Natur.363..558I. doi:10.1038/363558a0. PMID 8505985.
- Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L (September 2005). "A quantitative measurement of the human somatic mutation rate". Cancer Research. 65 (18): 8111–7. doi:10.1158/0008-5472.CAN-04-1198. PMID 16166284.
- "'Lifeless' prion proteins are 'capable of evolution'". Health. BBC News Online. London. 1 January 2010. Archived from the original on 25 September 2015. Retrieved 10 October 2015.
- Sullivan AD, Wigginton J, Kirschner D (August 2001). "The coreceptor mutation CCR5Delta32 influences the dynamics of HIV epidemics and is selected for by HIV". Proceedings of the National Academy of Sciences of the United States of America. 98 (18): 10214–9. Bibcode:2001PNAS...9810214S. doi:10.1073/pnas.181325198. PMC 56941. PMID 11517319.
- "Mystery of the Black Death". Secrets of the Dead. Season 3. Episode 2. 30 October 2002. PBS. Archived from the original on 12 October 2015. Retrieved 10 October 2015. Episode background.
- Galvani AP, Slatkin M (December 2003). "Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele". Proceedings of the National Academy of Sciences of the United States of America. 100 (25): 15276–9. Bibcode:2003PNAS..10015276G. doi:10.1073/pnas.2435085100. PMC 299980. PMID 14645720.
- Konotey-Ahulu, Felix. "Frequently Asked Questions [FAQ's]". sicklecell.md. Archived from the original on 30 April 2011.
- Hughes D, Andersson DI (September 2017). "Evolutionary Trajectories to Antibiotic Resistance". Annual Review of Microbiology. 71: 579–596. doi:10.1146/annurev-micro-090816-093813. PMID 28697667.
- Ségurel L, Bon C (August 2017). "On the Evolution of Lactase Persistence in Humans". Annual Review of Genomics and Human Genetics. 18: 297–319. doi:10.1146/annurev-genom-091416-035340. PMID 28426286.
- Bowler, Peter J. (1992) [1983]. The Eclipse of Darwinism. p. 198.
- Smocovitis, Vassiliki Betty (1996). Unifying Biology: The Evolutionary Synthesis and Evolutionary Biology. Journal of the History of Biology. 25. Princeton, NJ: Princeton University Press. p. 56. doi:10.1007/bf01947504. ISBN 978-0-691-03343-3. LCCN 96005605. OCLC 34411399. PMID 11623198.
- Hallgrímsson, Benedikt; Hall, Brian K. (2011). Variation: A Central Concept in Biology. Academic Press. p. 18.
- Sewall Wright. (1984). Evolution and the Genetics of Populations: Genetics and Biometric Foundations Volume 1. University of Chicago Press. p. 10
- De Vries, Hugo (1905). Species and Varieties: Their Origin by Mutation.
- Bateson, William (1894). Materials for the Study of Variation, Treated with Especial Regard to Discontinuity in the Origin of Species.
- Punnett, Reginald C. (1915). Mimicry in Butterflies. Cambridge University Press.
- Mayr, Ernst (2007). What Makes Biology Unique?: Considerations on the Autonomy of a Scientific Discipline. Cambridge University Press.
- Provine, W. B. (2001). The Origins of Theoretical Population Genetics, with a new afterword. University of Chicago Press, Chicago. pp. 56–107.
- Stoltzfus, A.; Cable, K. (2014). "Mendelian-Mutationism: The Forgotten Evolutionary Synthesis". J Hist Biol. 47 (4): 501–546. doi:10.1007/s10739-014-9383-2. PMID 24811736.
- Hull, D. L. (1985). "Darwinism as an historical entity: A historiographic proposal". In Kohn, D. (ed.). The Darwinian Heritage. Princeton University Press. pp. 773–812.
- Yule, G. Udny (1902). "Mendel's Laws and their probable relations to inter-racial heredity". New Phytologist. 1 (10): 226–227. doi:10.1111/j.1469-8137.1902.tb07336.x.
- Gould, S. J. (1982). "The uses of heresey; an introduction to Richard Goldschmidt's The Material Basis of Evolution." pp. xiii-xlii. Yale University Press.
- Ruse, Michael (1996). Monad to man: the Concept of Progress in Evolutionary Biology. Harvard University Press. pp. 412–413. ISBN 978-0-674-03248-4.
- A. Stoltzfus (2014). "In search of mutation-driven evolution". Evolution & Development. 16: 57–59. doi:10.1111/ede.12062.
- Futuyma, Douglas J. (2015). Serrelli, E.; Gontier, N. (eds.). Can Modern Evolutionary Theory Explain Macroevolution? (PDF). Macroevolution. Springer. pp. 29–85.
{kind=link}
External links
| Wikimedia Commons has media related to Mutations. |
- Jones S, Woolfson A, Partridge L (6 December 2007). "Genetic Mutation". In Our Time. BBC Radio 4. Retrieved 18 October 2015.
- Liou, Stephanie (5 February 2011). "All About Mutations". HOPES. Huntington's Disease Outreach Project for Education at Stanford. Retrieved 18 October 2015.
- "Locus Specific Mutation Databases". Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015.
- "Welcome to the Mutalyzer website". Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015. — The Mutalyzer website.
Evolutionary biology | ||
|---|---|---|
| ||
| Evolution |
|  |
| Population genetics | ||
| Development |
| |
| Of taxa | ||
| Of organs |
| |
| Of processes |
| |
| Tempo and modes |
| |
| Speciation |
| |
| History |
| |
| Philosophy |
| |
| Related |
| |
| ||