Ferroptosis
Ferroptosis is a type of programmed cell death dependent on iron and characterized by the accumulation of lipid peroxides, and is genetically and biochemically distinct from other forms of regulated cell death such as apoptosis.[1] Ferroptosis is initiated by the failure of the glutathione-dependent antioxidant defenses, resulting in unchecked lipid peroxidation and eventual cell death.[2] Lipophilic antioxidants and iron chelators can prevent ferroptotic cell death.
The accumulation of lipid peroxides that results in the process of ferroptosis makes it peroxidation-driven, meaning it is driven by the oxidative degeneration of lipids. This process of cell death occurs when free radical molecules take electrons from a lipid molecule to cause the degradation of the lipid molecule. This degradation is the result of the lipid molecules being oxidized, meaning losing electrons, to the free radical molecules. This process makes ferroptosis a regulatory form of cell death. The reason the process of oxidative degradation of a lipid drives ferroptosis is because glutathione peroxidase 4 (GPX4), a lipid repair enzyme, undergoes a loss of activity.[1] Researchers have identified roles in which ferroptosis can contribute to the medical field such as in aiding in cancer treatments where this form of cell death can be induced in the human body.[3] Ferroptosis activation plays a regulatory role on growth of tumor cells in the human body. However, the positive effects of ferroptosis could be potentially neutralized by its disruption of metabolic pathways and disruption of homeostasis in the human body.[4] Since ferroptosis is a form of regulated cell death, some of the molecules that regulate ferroptosis are involved in metabolic pathways that regulate cysteine exploitation, glutathione state, nicotinamide adenine dinucleotide phosphate function, lipid peroxidation and iron homeostasis.[4]
Mechanism of Ferroptosis
Small molecules such as Erastin, Sulfasalazine, Sorafenib, Altretamine, RSL-3, ML 162 and ML 210 are known inhibitors of this tumor cell growth and induce ferroptosis. They do not trigger a response of change in apoptosis and therefore have no chromatin margination or cleave poly ADP-ribose polymerase (PARP). Instead, the phenotype of the mitochondria is changed using primarily Erastin or RSL3. Iron is also a necessity for these activators. They therefore can be inhibited by iron chelators. Ultimately, cell death induced by Erastin and RSL-3 in the phenotype is what makes up ferroptosis. Ferroptosis can also be induced by blocking the enzyme GPX4. Triggering ferroptosis is also caused by inhibiting GSH, which is necessary for the function of GPX4, and ultimately inducing a ferroptotic response in a cell.[2]
Comparison To Apoptosis in the Nervous System
Another form of cell death that occurs in the nervous system is apoptosis. Named after the Greek word meaning “to fall away from”, apoptosis results in cell breakage into small, apoptotic bodies taken up through phagocytosis.[5] This process occurs continuously within mammalian nervous system processes that begin at fetal development and continue through adult life. Apoptotic death is crucial for the correct population size of neuronal and glial cells. Similarly to ferroptosis, deficiencies in apoptotic processes can result in many health complications, including neurodegeneration.
Within the study of neuronal apoptosis, most research has been conducted on the neurons of the superior cervical ganglion.[6] In order for these neurons to survive and innervate their target tissues, they must have nerve growth factor (NGF).[6] Normally, NGF binds to a tyrosine kinase receptor, TrkA, which activates phosphatidylinositol 3-kinase-Akt (PI3K-Akt) and extracellular signal-regulated kinase (Raf-MEK-ERK) signaling pathways. This occurs during normal development which promotes neuronal growth in the sympathetic nervous system.[6]
During embryonic development, the absence of NGF activates apoptosis by decreasing the activity of the signaling pathways normally activated by NGF.[6] this pathway of apoptosis has also been named the intrinsic pathway, as it is activated by intrinsic factors over external factors. Without NGF, the neurons of the sympathetic nervous system begin to atrophy, glucose uptake rates fall and the rates of protein synthesis and gene expression slow.[6] Apoptotic death from NGF withdrawal also requires caspase activity.[6] Upon NGF withdrawal, caspase-3 activation occurs through an in-vitro pathway beginning with the release of Cytochrome c from the mitochondria.[6] In a surviving sympathetic neuron, the overexpression of anti-apoptotic B-cell CLL/lymphoma 2 (Bcl-2) proteins prevents NGF withdrawal-induced death. However, overexpression of a separate, pro-apoptotic Bcl-2 gene, Bax, stimulates the release of Cytochrome c2. Cytochrome c promotes the activation of caspase-9 through the formation of the apoptosome. Once caspase-9 is activated, it can cleave and activate caspase-3 resulting in cell death. Notably, apoptosis does not release intracellular fluid as neurons that are degraded through ferroptosis do. During ferroptosis, neurons release lipid metabolites from inside the cell body. This is a key difference between ferroptosis and apoptosis.
Ferroptosis in Neurons

Neural connections are constantly changing within the nervous system. Synaptic connections that are used more often are kept intact and promoted, while synaptic connections that are rarely used are subject to degradation. Elevated levels of synaptic connection loss and degradation of neurons are linked to neurodegenerative diseases.[7] More recently, ferroptosis has been linked to these neurodegenerative diseases. Two new studies show that ferroptosis contributes to neuronal death after intracerebral hemorrhage.[8][9] Neurons that are degraded through ferroptosis release lipid metabolites from inside the cell body. The lipid metabolites are harmful to surrounding neurons, causing inflammation in the brain. Inflammation is a pathological feature of Alzheimer’s disease and intracerebral hemorrhage.
In a study performed using mice it was found that absence of a certain enzyme, glutathione peroxidase 4 (Gpx4), enhanced activity of ferroptosis. Foods high in Vitamin E promote Gpx4 activity, consequently inhibiting ferroptosis and preventing inflammation in brain regions. In the experimental group of mice that were manipulated to have decreased Gpx4 levels, mice were observed to have cognitive impairment and neurodegeneration of hippocampal neurons, again linking ferroptosis to neurodegenerative diseases.
Similarly, presence of transcription factors, specifically ATF-4, can determine how readily a neuron can undergo cell death. The presence of ATF-4 promotes resistance in cells against ferroptosis. However, this resistance can cause other diseases, such as cancer, to progress and become malignant. While ATF-4 provides resistance ferroptosis, an abundance of ATF-4 causes neurodegeneration.
Role of Ferroptosis in Cancer Treatment
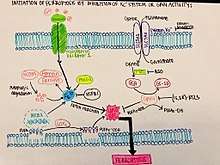
Ferroptosis is a more controlled form of programmed cell death. With small molecules inducing ferroptosis, it inhibits tumor growth and increases drug resistance. It is still unknown the exact mechanism in which ferroptosis inhibits tumor growth, but based on early experiments, it could be used to treat tumor cells and ultimately cancer. Based on the figure shown, initiating ferroptosis can be done by eliminating GPX4 activity through Xc- (erastin mediated). The inhibition or degradation of GPX4 by eratin or RSL3 has been noticed to be the primary mechanism that will trigger ferroptosis. Since ferroptosis is an iron dependent mechanism, an accumulation of lipid ROS kills the cells that have begun to undergo ferroptosis and eliminates them completely.
Ferroptosis can be used to treat several different types of cancer, each seemingly different forms of the disease. This method of cell death has been tested either in mice, or is in the early stages of research and has not yet been fully tested. This includes cancer types such as:
- Breast
- Acute myeloid leukemia
- Pancreatic ductal adenocarcinoma
- Ovarian
- B cell lymphoma
- Renal cell carcinomas
- Lung
- Glioblastoma
These forms of cancer have been hypothesized to be highly sensitive to ferroptosis, or react to Erastin or Xc- in a way that minimizes the tumor cells or responds to this treatment with a result of cell death. An upregulation of iron levels has also been seen to induce ferroptosis in certain types of cancer, such as breast cancer.[3] Breast cancer cells have exhibited vulnerability to ferroptosis via a combination of siramesine and lapatinib. These cells also exhibited an autophagic cycle independent of ferroptotic activity, indicating that the two different forms of cell death could be controlled to activate at specific times following treatment.[10]
See also
References
- 1 2 Yang, Wan; Stockwell, Bent (2 December 2015). "Ferroptosis: death by lipid peroxidation". PubMed Central. doi:10.1016/j.tcb.2015.10.014. PMC 4764384. PMID 26653790.
- 1 2 Cao, Jennifer; Dixon, Scott (5 April 2016). "Mechanisms of ferroptosis". PubMed Central. doi:10.1007/s00018-016-2914-1. PMID 27048822.
- 1 2 Lu, Bin; Chen, Xiao; Ying, Mei; He, Quiao; Cao, Ji; Yang, Bo (12 January 2018). "The Role of Ferroptosis in Cancer Development and Treatment Response". PubMed Central. doi:10.3389/fphar.2017.00992. PMID 29375387.
- 1 2 Hao, S; Liang, B; Huang, Q; Dong, S; Wu, Z; He, W; Shi, M (15 February 2018). "Metabolic Networks in Ferroptosis". PubMed Central. doi:10.3892/ol.2018.8066. PMID 29556292.
- ↑ Reed, John (November 2000). "Mechanisms of Apoptosis". The American Journal of Pathology. 157 (5): 1415–1430. doi:10.1016/S0002-9440(10)64779-7. PMC 1885741.
- 1 2 3 4 5 6 7 Kristiansen, M; Ham, J (25 April 2014). "Programmed cell death during neuronal development: the sympathetic neuron model". PubMed Central. doi:10.1038/cdd.2014.47. PMC 4207485. PMID 24769728.
- ↑ Hambright, WS; Fonseca, RS; Chen, L; Na, R; Ran, Q (1 February 2017). "Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration". PubMed Central. doi:10.1016/j.redox.2017.01.021. PMID 28212525.
- ↑ Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, Ying M, Koehler RC, Stockwell BR, Wang J (April 2017). "Inhibition of neuronal ferroptosis protects hemorrhagic brain". JCI Insight. 2 (7): e90777. doi:10.1172/jci.insight.90777. PMC 5374066. PMID 27143417.
- ↑ Li Q, Weiland A, Chen X, et al. (July 2018). "Ultrastructural Characteristics of Neuronal Death and White Matter Injury in Mouse Brain Tissues After Intracerebral Hemorrhage: Coexistence of Ferroptosis, Autophagy, and Necrosis". Front Neurol. 9: 581. doi:10.3389/fneur.2018.00581. PMC 6056664. PMID 30065697.
- ↑ Ma, Shumei, Rebecca F. Dielschneider, Elizabeth S. Henson, Wenyan Xiao, Tricia R. Choquette, Anna R. Blankstein, Yongqiang Chen, and Spencer B. Gibson. “Ferroptosis and Autophagy Induced Cell Death Occur Independently after Siramesine and Lapatinib Treatment in Breast Cancer Cells.” PLOS ONE 12, no. 8 (August 21, 2017): e0182921. https://doi.org/10.1371/journal.pone.0182921.