Iron
 | |||||||||||||||||||||||||||||||||||||||||
| General properties | |||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Appearance | lustrous metallic with a grayish tinge | ||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight (Ar, standard) | 55.845(2)[1] | ||||||||||||||||||||||||||||||||||||||||
| Iron in the periodic table | |||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 26 | ||||||||||||||||||||||||||||||||||||||||
| Group | group 8 | ||||||||||||||||||||||||||||||||||||||||
| Period | period 4 | ||||||||||||||||||||||||||||||||||||||||
| Element category | transition metal | ||||||||||||||||||||||||||||||||||||||||
| Block | d-block | ||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Ar] 3d6 4s2 | ||||||||||||||||||||||||||||||||||||||||
Electrons per shell | 2, 8, 14, 2 | ||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | ||||||||||||||||||||||||||||||||||||||||
| Melting point | 1811 K (1538 °C, 2800 °F) | ||||||||||||||||||||||||||||||||||||||||
| Boiling point | 3134 K (2862 °C, 5182 °F) | ||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 7.874 g/cm3 | ||||||||||||||||||||||||||||||||||||||||
| when liquid (at m.p.) | 6.98 g/cm3 | ||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | 13.81 kJ/mol | ||||||||||||||||||||||||||||||||||||||||
| Heat of vaporization | 340 kJ/mol | ||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | 25.10 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||
Vapor pressure
| |||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||
| Oxidation states |
−4, −2, −1, +1,[2] +2, +3, +4, +5,[3] +6, +7[4] | ||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.83 | ||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
| ||||||||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 126 pm | ||||||||||||||||||||||||||||||||||||||||
| Covalent radius |
Low spin: 132±3 pm High spin: 152±6 pm | ||||||||||||||||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||
| Crystal structure |
body-centered cubic (bcc) a=286.65 pm | ||||||||||||||||||||||||||||||||||||||||
| Crystal structure |
face-centered cubic (fcc) between 1185–1667 K | ||||||||||||||||||||||||||||||||||||||||
| Speed of sound thin rod | 5120 m/s (at r.t.) (electrolytic) | ||||||||||||||||||||||||||||||||||||||||
| Thermal expansion | 11.8 µm/(m·K) (at 25 °C) | ||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 80.4 W/(m·K) | ||||||||||||||||||||||||||||||||||||||||
| Electrical resistivity | 96.1 nΩ·m (at 20 °C) | ||||||||||||||||||||||||||||||||||||||||
| Curie point | 1043 K | ||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | ferromagnetic | ||||||||||||||||||||||||||||||||||||||||
| Young's modulus | 211 GPa | ||||||||||||||||||||||||||||||||||||||||
| Shear modulus | 82 GPa | ||||||||||||||||||||||||||||||||||||||||
| Bulk modulus | 170 GPa | ||||||||||||||||||||||||||||||||||||||||
| Poisson ratio | 0.29 | ||||||||||||||||||||||||||||||||||||||||
| Mohs hardness | 4 | ||||||||||||||||||||||||||||||||||||||||
| Vickers hardness | 608 MPa | ||||||||||||||||||||||||||||||||||||||||
| Brinell hardness | 200–1180 MPa | ||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7439-89-6 | ||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||
| Discovery | before 5000 BC | ||||||||||||||||||||||||||||||||||||||||
| Main isotopes of iron | |||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||
Iron is a chemical element with symbol Fe (from Latin: ferrum) and atomic number 26. It is a metal in the first transition series. It is by mass the most common element on Earth, forming much of Earth's outer and inner core. It is the fourth most common element in the Earth's crust. Its abundance in rocky planets like Earth is due to its abundant production by fusion in high-mass stars, where it is the last element to be produced with release of energy before the violent collapse of a supernova, which scatters the iron into space.
Like the other group 8 elements, ruthenium and osmium, iron exists in a wide range of oxidation states, −2 to +7, although +2 and +3 are the most common. Elemental iron occurs in meteoroids and other low oxygen environments, but is reactive to oxygen and water. Fresh iron surfaces appear lustrous silvery-gray, but oxidize in normal air to give hydrated iron oxides, commonly known as rust. Unlike the metals that form passivating oxide layers, iron oxides occupy more volume than the metal and thus flake off, exposing fresh surfaces for corrosion.
Iron metal has been used since ancient times, although copper alloys, which have lower melting temperatures, were used even earlier in human history. Pure iron is relatively soft, but is unobtainable by smelting because it is significantly hardened and strengthened by impurities, in particular carbon, from the smelting process. A certain proportion of carbon (between 0.002% and 2.1%) produces steel, which may be up to 1000 times harder than pure iron. Crude iron metal is produced in blast furnaces, where ore is reduced by coke to pig iron, which has a high carbon content. Further refinement with oxygen reduces the carbon content to the correct proportion to make steel. Steels and iron alloys formed with other metals (alloy steels) are by far the most common industrial metals because they have a great range of desirable properties and iron-bearing rock is abundant.
Iron chemical compounds have many uses. Iron oxide mixed with aluminium powder can be ignited to create a thermite reaction, used in welding and purifying ores. Iron forms binary compounds with the halogens and the chalcogens. Among its organometallic compounds is ferrocene, the first sandwich compound discovered.
Iron plays an important role in biology, forming complexes with molecular oxygen in hemoglobin and myoglobin; these two compounds are common oxygen transport proteins in vertebrates. Iron is also the metal at the active site of many important redox enzymes dealing with cellular respiration and oxidation and reduction in plants and animals. In adult human males are some 3.8 grams of iron, and 2.3 grams in females, for whom iron is distributed in hemoglobin and throughout the body. Iron is a critical element in the metabolism of hundreds of proteins and enzymes involved in diverse body functions, such as oxygen transport, DNA synthesis, and cell growth.[5]
Characteristics
Mechanical properties
| Material | TS (MPa) |
BH (Brinell) |
|---|---|---|
| Iron whiskers | 11000 | |
| Ausformed (hardened) steel |
2930 | 850–1200 |
| Martensitic steel | 2070 | 600 |
| Bainitic steel | 1380 | 400 |
| Pearlitic steel | 1200 | 350 |
| Cold-worked iron | 690 | 200 |
| Small-grain iron | 340 | 100 |
| Carbon-containing iron | 140 | 40 |
| Pure, single-crystal iron | 10 | 3 |
The mechanical properties of iron and its alloys can be evaluated using a variety of tests, including the Brinell test, Rockwell test and the Vickers hardness test. The data on iron is so consistent that it is often used to calibrate measurements or to compare tests.[7][8] However, the mechanical properties of iron are significantly affected by the sample's purity: pure, single crystals of iron are actually softer than aluminium,[6] and the purest industrially produced iron (99.99%) has a hardness of 20–30 Brinell.[9] An increase in the carbon content will cause a significant increase in the hardness and tensile strength of iron. Maximum hardness of 65 Rc is achieved with a 0.6% carbon content, although the alloy has low tensile strength.[10] Because of the softness of iron, it is much easier to work with than its heavier congeners ruthenium and osmium.[11]
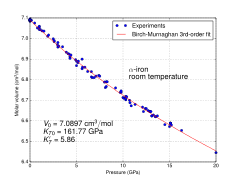
Because of its significance for planetary cores, the physical properties of iron at high pressures and temperatures have also been studied extensively. The form of iron that is stable under standard conditions can be subjected to pressures up to ca. 15 GPa before transforming into a high-pressure form, as described in the next section.
Phase diagram and allotropes
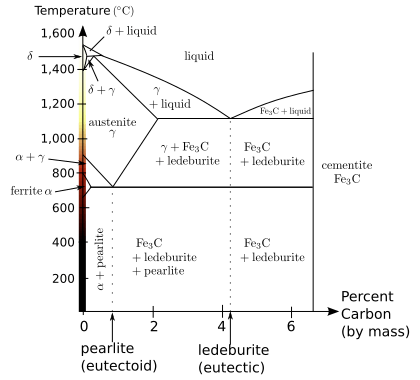
Iron represents an example of allotropy in a metal. At least four allotropic forms of iron are known as α, γ, δ, and ε; at very high pressures and temperatures, some controversial experimental evidence exists for a stable β phase.[12]
.png)
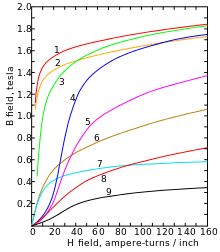
As molten iron cools past its freezing point of 1538 °C, it crystallizes into its δ allotrope, which has a body-centered cubic (bcc) crystal structure. As it cools further to 1394 °C, it changes to its γ-iron allotrope, a face-centered cubic (fcc) crystal structure, or austenite. At 912 °C and below, the crystal structure again becomes the bcc α-iron allotrope. Finally, at 770 °C (the Curie point, Tc) iron's magnetic ordering changes from paramagnetic to ferromagnetic. As is passes through the Curie temperature, iron does not change its structure, but "magnetic domains" appear, where each domain contains iron atoms with a particular electronic spin. In unmagnetized iron, all the electronic spins of the atoms within one domain have the same axis orientation; however, the electrons of neighboring domains have other orientations with the result of mutual cancellation and no magnetic field. In magnetized iron, the electronic spins of the domains are aligned and the magnetic effects are reinforced. Although each domain contains billions of atoms, they are very small, about 10 micrometres across.[14] This happens because the two unpaired electrons on each iron atom are in the dz2 and dx2 − y2 orbitals, which do not point directly at the nearest neighbors in the body-centered cubic lattice and therefore do not participate in metallic bonding; thus, they can interact magnetically with each other so that their spins align.[15]
At pressures above approximately 10 GPa and temperatures of a few hundred kelvin or less, α-iron changes into a hexagonal close-packed (hcp) structure, which is also known as ε-iron; the higher-temperature γ-phase also changes into ε-iron, but does so at higher pressure. The β-phase, if it exists, would appear at pressures of at least 50 GPa and temperatures of at least 1500 K and have an orthorhombic or a double hcp structure.[12] These high-pressure phases of iron are important as endmember models for the solid parts of planetary cores. The inner core of the Earth is generally presumed to be an iron-nickel alloy with ε (or β) structure.[16] Somewhat confusingly, the term "β-iron" is sometimes also used to refer to α-iron above its Curie point, when it changes from being ferromagnetic to paramagnetic, even though its crystal structure has not changed.[15]
The melting point of iron is experimentally well defined for pressures less than 50 GPa. For greater pressures, studies put the γ-ε-liquid triple point at pressures that differ by tens of gigapascals and 1000 K in the melting point. Generally speaking, molecular dynamics computer simulations of iron melting and shock wave experiments suggest higher melting points and a much steeper slope of the melting curve than static experiments carried out in diamond anvil cells.[17] The melting and boiling points of iron, along with its enthalpy of atomization, are lower than those of the earlier 3d elements from scandium to chromium, showing the lessened contribution of the 3d electrons to metallic bonding as they are attracted more and more into the inert core by the nucleus;[18] however, they are higher than the values for the previous element manganese because that element has a half-filled 3d subshell and consequently its d-electrons are not easily delocalized. This same trend appears for ruthenium but not osmium.[11]
Isotopes
Naturally occurring iron consists of four stable isotopes: 5.845% of 54Fe, 91.754% of 56Fe, 2.119% of 57Fe and 0.282% of 58Fe. Of these stable isotopes, only 57Fe has a nuclear spin (−1⁄2). The nuclide 54Fe theoretically can undergo double electron capture to 54Cr, but the process has never been observed and only a lower limit on the half-life of 3.1×1022 years has been established.[19]
60Fe is an extinct radionuclide of long half-life (2.6 million years).[20] It is not found on Earth, but its ultimate decay product is its granddaughter, the stable nuclide 60Ni.[19] Much of the past work on isotopic composition of iron has focused on the nucleosynthesis of 60Fe through studies of meteorites and ore formation. In the last decade, advances in mass spectrometry have allowed the detection and quantification of minute, naturally occurring variations in the ratios of the stable isotopes of iron. Much of this work is driven by the Earth and planetary science communities, although applications to biological and industrial systems are emerging.[21]
In phases of the meteorites Semarkona and Chervony Kut, a correlation between the concentration of 60Ni, the granddaughter of 60Fe, and the abundance of the stable iron isotopes provided evidence for the existence of 60Fe at the time of formation of the Solar System. Possibly the energy released by the decay of 60Fe, along with that released by 26Al, contributed to the remelting and differentiation of asteroids after their formation 4.6 billion years ago. The abundance of 60Ni present in extraterrestrial material may bring further insight into the origin and early history of the Solar System.[22]
The most abundant iron isotope 56Fe is of particular interest to nuclear scientists because it represents the most common endpoint of nucleosynthesis.[23] Since 56Ni (14 alpha particles) is easily produced from lighter nuclei in the alpha process in nuclear reactions in supernovae (see silicon burning process), it is the endpoint of fusion chains inside extremely massive stars, since addition of another alpha particle, resulting in 60Zn, requires a great deal more energy. This 56Ni, which has a half-life of about 6 days, is created in quantity in these stars, but soon decays by two successive positron emissions within supernova decay products in the supernova remnant gas cloud, first to radioactive 56Co, and then to stable 56Fe. As such, iron is the most abundant element in the core of red giants, and is the most abundant metal in iron meteorites and in the dense metal cores of planets such as Earth.[24] It is also very common in the universe, relative to other stable metals of approximately the same atomic weight.[24][25] Iron is the sixth most abundant element in the Universe, and the most common refractory element.[26]
Although a further tiny energy gain could be extracted by synthesizing 62Ni, which has a marginally higher binding energy than 56Fe, conditions in stars are unsuitable for this process. Element production in supernovas and distribution on Earth greatly favor iron over nickel, and in any case, 56Fe still has a lower mass per nucleon than 62Ni due to its higher fraction of lighter protons.[27] Hence, elements heavier than iron require a supernova for their formation, involving rapid neutron capture by starting 56Fe nuclei.[24]
In the far future of the universe, assuming that proton decay does not occur, cold fusion occurring via quantum tunnelling would cause the light nuclei in ordinary matter to fuse into 56Fe nuclei. Fission and alpha-particle emission would then make heavy nuclei decay into iron, converting all stellar-mass objects to cold spheres of pure iron.[28]

Occurrence

Metallic or native iron is rarely found on the surface of the Earth because it tends to oxidize, but its oxides are pervasive and represent the primary ores. While it makes up about 5% of the Earth's crust, both the Earth's inner and outer core are believed to consist largely of an iron-nickel alloy constituting 35% of the mass of the Earth as a whole. Iron is consequently the most abundant element on Earth, but only the fourth most abundant element in the Earth's crust, after oxygen, silicon, and aluminium.[29] Most of the iron in the crust is found combined with oxygen as iron oxide minerals such as hematite (Fe2O3), magnetite (Fe3O4), and siderite (FeCO3). Many igneous rocks also contain the sulfide minerals pyrrhotite and pentlandite.[30][31]
Ferropericlase (Mg,Fe)O, a solid solution of periclase (MgO) and wüstite (FeO), makes up about 20% of the volume of the lower mantle of the Earth, which makes it the second most abundant mineral phase in that region after silicate perovskite (Mg,Fe)SiO3; it also is the major host for iron in the lower mantle.[32] At the bottom of the transition zone of the mantle, the reaction γ-(Mg,Fe)2[SiO4] ↔ (Mg,Fe)[SiO3] + (Mg,Fe)O transforms γ-olivine into a mixture of perovskite and ferropericlase and vice versa. In the literature, this mineral phase of the lower mantle is also often called magnesiowüstite.[33] Silicate perovskite may form up to 93% of the lower mantle,[34] and the magnesium iron form, (Mg,Fe)SiO3, is considered to be the most abundant mineral in the Earth, making up 38% of its volume.[35]
Large deposits of iron are found in banded iron formations. These geological formations are a type of rock consisting of repeated thin layers of iron oxides alternating with bands of iron-poor shale and chert. The banded iron formations were laid down in the time between 3,700 million years ago and 1,800 million years ago.[36][37]
The mentioned iron compounds have been used as pigments (compare ochre) since historical time and contribute as well to the color of various geological formations, e.g. the Buntsandstein (British Bunter, colored sandstein).[38] In the case of the Eisensandstein (a jurassic 'iron sandstone', e.g. from Donzdorf) in Germany[39] and Bath stone in the UK, iron pigments contribute to the yellowish color of large amounts of historical buildings and sculptures.[40] The proverbial red color of the surface of Mars is derived from an iron oxide-rich regolith.[41]
Significant amounts of iron occur in the iron sulfide mineral pyrite (FeS2), but it is difficult to extract iron from it and it is therefore not used. In fact, iron is so common that production generally focuses only on ores with very high quantities of it. During weathering, iron tends to leach from sulfide deposits as the sulfate and from silicate deposits as the bicarbonate. Both of these are oxidized in aqueous solution and precipitate in even mildly elevated pH as iron(III) oxide.[42]
About 1 in 20 meteorites consist of the unique iron-nickel minerals taenite (35–80% iron) and kamacite (90–95% iron). Although rare, iron meteorites are the main form of natural metallic iron on the Earth's surface.[43] According to the International Resource Panel's Metal Stocks in Society report, the global stock of iron in use in society is 2200 kg per capita. More-developed countries differ in this respect from less-developed countries (7000–14000 vs 2000 kg per capita).[44]
Chemistry and compounds
| Oxidation state | Representative compound |
|---|---|
| −2 (d10) | Disodium tetracarbonylferrate (Collman's reagent) |
| −1 (d9) | Fe 2(CO)2− 8 |
| 0 (d8) | Iron pentacarbonyl |
| 1 (d7) | Cyclopentadienyliron dicarbonyl dimer ("Fp2") |
| 2 (d6) | Ferrous sulfate, ferrocene |
| 3 (d5) | Ferric chloride, ferrocenium tetrafluoroborate |
| 4 (d4) | Fe(diars) 2Cl2+ 2 |
| 5 (d3) | FeO3− 4 |
| 6 (d2) | Potassium ferrate |
| 7 (d1) | [FeO4]– (matrix isolation, 4K) |
Iron shows the characteristic chemical properties of the transition metals, namely the ability to form variable oxidation states differing by steps of one and a very large coordination and organometallic chemistry: indeed, it was the discovery of an iron compound, ferrocene, that revolutionalized the latter field in the 1950s.[45] Iron is sometimes considered as a prototype for the entire block of transition metals, due to its abundance and the immense role it has played in the technological progress of humanity.[46] Its 26 electrons are arranged in the configuration [Ar]3d64s2, of which the 3d and 4s electrons are relatively close in energy, and thus it can lose a variable number of electrons and there is no clear point where further ionization becomes unprofitable.[11]
Iron forms compounds mainly in the +2 and +3 oxidation states. Traditionally, iron(II) compounds are called ferrous, and iron(III) compounds ferric. Iron also occurs in higher oxidation states, e.g. the purple potassium ferrate (K2FeO4), which contains iron in its +6 oxidation state. Although iron(VIII) oxide (FeO4) has been claimed, the report could not be reproduced and such a species (at least with iron in its +8 oxidation state) has been found to be improbable computationally.[47] However, one form of anionic [FeO4]– with iron in its +7 oxidation state, along with an iron(V)-peroxo isomer, has been detected by infrared spectroscopy at 4 K after cocondensation of laser-ablated Fe atoms with a mixture of O2/Ar.[48] Iron(IV) is a common intermediate in many biochemical oxidation reactions.[49][50] Numerous organoiron compounds contain formal oxidation states of +1, 0, −1, or even −2. The oxidation states and other bonding properties are often assessed using the technique of Mössbauer spectroscopy.[51] Many mixed valence compounds contain both iron(II) and iron(III) centers, such as magnetite and Prussian blue (Fe4(Fe[CN]6)3).[50] The latter is used as the traditional "blue" in blueprints.[52]
Iron is the first of the transition metals that cannot reach its group oxidation state of +8, although its heavier congeners ruthenium and osmium can, with ruthenium having more difficulty than osmium.[15] Ruthenium exhibits an aqueous cationic chemistry in its low oxidation states similar to that of iron, but osmium does not, favoring high oxidation states in which it forms anionic complexes.[15] In the second half of the 3d transition series, vertical similarities down the groups compete with the horizontal similarities of iron with its neighbors cobalt and nickel in the periodic table, which are also ferromagnetic at room temperature and share similar chemistry. As such, iron, cobalt, and nickel are sometimes grouped together as the iron triad.[46]
_chloride_hexahydrate.jpg)
The iron compounds produced on the largest scale in industry are iron(II) sulfate (FeSO4·7H2O) and iron(III) chloride (FeCl3). The former is one of the most readily available sources of iron(II), but is less stable to aerial oxidation than Mohr's salt ((NH4)2Fe(SO4)2·6H2O). Iron(II) compounds tend to be oxidized to iron(III) compounds in the air.[50]
Unlike many other metals, iron does not form amalgams with mercury. As a result, mercury is traded in standardized 76 pound flasks (34 kg) made of iron.[53]
Iron is by far the most reactive element in its group; it is pyrophoric when finely divided and dissolves easily in dilute acids, giving Fe2+. However, it does not react with concentrated nitric acid and other oxidizing acids due to the formation of an impervious oxide layer, which can nevertheless react with hydrochloric acid.[15]
Binary compounds
Iron reacts with oxygen in the air to form various oxide and hydroxide compounds; the most common are iron(II,III) oxide (Fe3O4), and iron(III) oxide (Fe2O3). Iron(II) oxide also exists, though it is unstable at room temperature. Despite their names, they are actually all non-stoichiometric compounds whose compositions may vary.[54] These oxides are the principal ores for the production of iron (see bloomery and blast furnace). They are also used in the production of ferrites, useful magnetic storage media in computers, and pigments. The best known sulfide is iron pyrite (FeS2), also known as fool's gold owing to its golden luster.[50] It is not an iron(IV) compound, but is actually an iron(II) polysulfide containing Fe2+ and S2−
2 ions in a distorted sodium chloride structure.[54]
The binary ferrous and ferric halides are well-known, with the exception of ferric iodide. The ferrous halides typically arise from treating iron metal with the corresponding hydrohalic acid to give the corresponding hydrated salts.[50]
- Fe + 2 HX → FeX2 + H2 (X = F, Cl, Br, I)
Iron reacts with fluorine, chlorine, and bromine to give the corresponding ferric halides, ferric chloride being the most common.[55]
- 2 Fe + 3 X2 → 2 FeX3 (X = F, Cl, Br)
Ferric iodide is an exception, being thermodynamically unstable due to the oxidizing power of Fe3+ and the high reducing power of I−:[55]
- 2 I− + 2 Fe3+ → I2 + 2 Fe2+ (E0 = +0.23 V)
Nevertheless, milligram amounts of ferric iodide, a black solid, may still be prepared through the reaction of iron pentacarbonyl with iodine and carbon monoxide in the presence of hexane and light at the temperature of −20 °C, making sure that the system is well sealed off from air and water.[55]
Solution chemistry

The standard reduction potentials in acidic aqueous solution for some common iron ions are given below:[15]
| Fe2+ + 2 e− | ⇌ Fe | E0 = −0.447 V |
| Fe3+ + 3 e− | ⇌ Fe | E0 = −0.037 V |
| FeO2− 4 + 8 H+ + 3 e− | ⇌ Fe3+ + 4 H2O | E0 = +2.20 V |
The red-purple tetrahedral ferrate(VI) anion is such a strong oxidizing agent that it oxidizes nitrogen and ammonia at room temperature, and even water itself in acidic or neutral solutions:[55]
- 4 FeO2−
4 + 10 H
2O → 4 Fe3+
+ 20 OH−
+ 3 O2
The Fe3+ ion has a large simple cationic chemistry, although the pale-violet hexaquo ion [Fe(H2O)6]3+ is very readily hydrolyzed when pH increases above 0 as follows:[56]
| [Fe(H2O)6]3+ | ⇌ [Fe(H2O)5(OH)]2+ + H+ | K = 10−3.05 mol dm−3 |
| [Fe(H2O)5(OH)]2+ | ⇌ [Fe(H2O)4(OH)2]+ + H+ | K = 10−3.26 mol dm−3 |
| 2 [Fe(H2O)6]3+ | ⇌ [Fe(H 2O) 4(OH)]4+ 2 + 2 H+ + 2 H2O | K = 10−2.91 mol dm−3 |
-oxide-sample.jpg)
-sulfate-heptahydrate-sample.jpg)
As pH rises above 0 the above yellow hydrolyzed species form and as it rises above 2–3, reddish-brown hydrous iron(III) oxide precipitates out of solution. Although Fe3+ has an d5 configuration, its absorption spectrum is not like that of Mn2+ with its weak, spin-forbidden d–d bands, because Fe3+ has higher positive charge and is more polarizing, lowering the energy of its ligand-to-metal charge transfer absorptions. Thus, all the above complexes are rather strongly colored, with the single exception of the hexaquo ion – and even that has a spectrum dominated by charge transfer in the near ultraviolet region.[56] On the other hand, the pale green iron(II) hexaquo ion [Fe(H2O)6]2+ does not undergo appreciable hydrolysis. Carbon dioxide is not evolved when carbonate anions are added, which instead results in white iron(II) carbonate being precipitated out. In excess carbon dioxide this forms the slightly soluble bicarbonate, which occurs commonly in groundwater, but it oxidises quickly in air to form iron(III) oxide that accounts for the brown deposits present in a sizeable number of streams.[57]
Coordination compounds
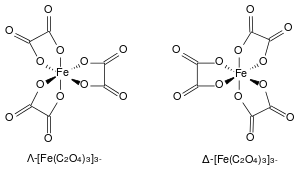
Many coordination compounds of iron are known. A typical six-coordinate anion is hexachloroferrate(III), [FeCl6]3−, found in the mixed salt tetrakis(methylammonium) hexachloroferrate(III) chloride.[58][59] Complexes with multiple bidentate ligands have geometric isomers. For example, the trans-chlorohydridobis(bis-1,2-(diphenylphosphino)ethane)iron(II) complex is used as a starting material for compounds with the Fe(dppe)2 moiety.[60][61] The ferrioxalate ion with three oxalate ligands (shown at right) displays helical chirality with its two non-superposable geometries labelled Λ (lambda) for the left-handed screw axis and Δ (delta) for the right-handed screw axis, in line with IUPAC conventions.[56] Potassium ferrioxalate is used in chemical actinometry and along with its sodium salt undergoes photoreduction applied in old-style photographic processes. The dihydrate of iron(II) oxalate has a polymeric structure with co-planar oxalate ions bridging between iron centres with the water of crystallisation located forming the caps of each octahedron, as illustrated below.[62]
(H2O)2-chain-from-xtal-2008-CM-3D-balls.png)
Prussian blue, Fe4[Fe(CN)6]3, is the most famous of the cyanide complexes of iron. Its formation can be used as a simple wet chemistry test to distinguish between aqueous solutions of Fe2+ and Fe3+ as they react (respectively) with potassium ferricyanide and potassium ferrocyanide to form Prussian blue.[50]
iron(III)_chloride.jpg)
Iron(III) complexes are quite similar to those of chromium(III) with the exception of iron(III)'s preference for O-donor instead of N-donor ligands. The latter tend to be rather more unstable than iron(II) complexes and often dissociate in water. Many Fe–O complexes show intense colors and are used as tests for phenols or enols. For example, in the ferric chloride test, used to determine the presence of phenols, iron(III) chloride reacts with a phenol to form a deep violet complex:[56]
- 3 ArOH + FeCl3 → Fe(OAr)3 + 3 HCl (Ar = aryl)
Among the halide and pseudohalide complexes, fluoro complexes of iron(III) are the most stable, with the colorless [FeF5(H2O)]2− being the most stable in aqueous solution. Chloro complexes are less stable and favor tetrahedral coordination as in [FeCl4]−; [FeBr4]− and [FeI4]− are reduced easily to iron(II). Thiocyanate is a common test for the presence of iron(III) as it forms the blood-red [Fe(SCN)(H2O)5]2+. Like manganese(II), most iron(III) complexes are high-spin, the exceptions being those with ligands that are high in the spectrochemical series such as cyanide. An example of a low-spin iron(III) complex is [Fe(CN)6]3−. The cyanide ligands may easily be detached in [Fe(CN)6]3−, and hence this complex is poisonous, unlike the iron(II) complex [Fe(CN)6]4− found in Prussian blue,[56] which does not release hydrogen cyanide except when dilute acids are added.[57] Iron shows a great variety of electronic spin states, including every possible spin quantum number value for a d-block element from 0 (diamagnetic) to 5⁄2 (5 unpaired electrons). This value is always half the number of unpaired electrons. Complexes with zero to two unpaired electrons are considered low-spin and those with four or five are considered high-spin.[54]
Iron(II) complexes are less stable than iron(III) complexes but the preference for O-donor ligands is less marked, so that for example [Fe(NH3)6]2+ is known while [Fe(NH3)6]3+ is not. They have a tendency to be oxidized to iron(III) but this can be moderated by low pH and the specific ligands used.[57]
Organometallic compounds

.JPG)
Cyanide complexes are technically organometallic but more important are carbonyl complexes and sandwich and half-sandwich compounds. The premier iron(0) compound is iron pentacarbonyl, Fe(CO)5, which is used to produce carbonyl iron powder, a highly reactive form of metallic iron. Thermolysis of iron pentacarbonyl gives the trinuclear cluster, triiron dodecacarbonyl. Collman's reagent, disodium tetracarbonylferrate, is a useful reagent for organic chemistry; it contains iron in the −2 oxidation state. Cyclopentadienyliron dicarbonyl dimer contains iron in the rare +1 oxidation state.[63]
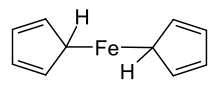
Ferrocene was an extremely important compound in the early history of the branch of organometallic chemistry, and to this day iron is still one of the most important metals in this field.[64] It was first synthesised in 1951 during an attempt to prepare the fulvalene (C10H8) by oxidative dimerization of cyclopentadiene; the resultant product was found to have molecular formula C10H10Fe and reported to exhibit "remarkable stability".[65] The discovery sparked substantial interest in the field of organometallic chemistry,[66][67] in part because the structure proposed by Pauson and Kealy (shown at right) was inconsistent with then-existing bonding models and did not explain its unexpected stability. Consequently, the initial challenge was to definitively determine the structure of ferrocene in the hope that its bonding and properties would then be understood. The shockingly novel sandwich structure, [Fe(η5-C5H5)2],[66] was deduced and reported independently by three groups in 1952: Robert Burns Woodward and Geoffrey Wilkinson investigated the reactivity in order to determine the structure[68] and demonstrated that ferrocene undergoes similar reactions to a typical aromatic molecule (such as benzene),[69] Ernst Otto Fischer deduced the sandwich structure and also began synthesising other metallocenes including cobaltocene;[70] Eiland and Pepinsky provided X-ray crystallographic confirmation of the sandwich structure.[71]
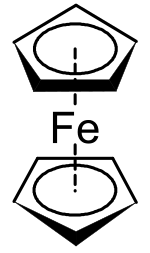
Applying valence bond theory to ferrocene by considering an Fe2+ centre and two cyclopentadienide anions (C5H5−), which are known to be aromatic according to Hückel's rule and hence highly stable, allowed correct prediction of the geometry of the molecule. Once molecular orbital theory was successfully applied and the Dewar-Chatt-Duncanson model proposed,[72] the reasons for ferrocene's remarkable stability became clear.[73] Ferrocene was not the first organometallic compound known – Zeise's salt, K[PtCl3(C2H4)]·H2O was reported in 1831[74][75] and Mond's discovery of Ni(CO)4 occurred in 1888,[76] but it was ferrocene's discovery that began organometallic chemistry as a separate area of chemistry. It was so important that Wilkinson and Fischer shared the 1973 Nobel Prize for Chemistry "for their pioneering work, performed independently, on the chemistry of the organometallic, so called sandwich compounds".[77] Ferrocene itself can be used as the backbone of a ligand, e.g. 1,1'-bis(diphenylphosphino)ferrocene (dppf). Ferrocene can itself be oxidized to the ferrocenium cation (Fc+); the ferrocene/ferrocenium couple is often used as a reference in electrochemistry.[67]
Metallocenes like ferrocene can be prepared by reaction of freshly-cracked cyclopentadiene with iron(II) chloride and base.[78] It is an aromatic substance and undergoes substitution reactions rather than addition reactions on the cyclopentadienyl ligands. For example, Friedel-Crafts acylation of ferrocene with acetic anhydride yields acetylferrocene[79] just as acylation of benzene yields acetophenone under similar conditions.
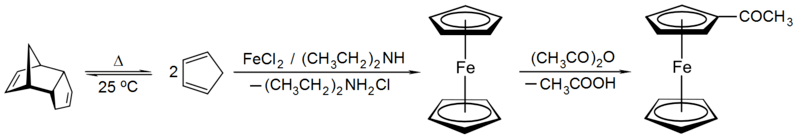
Iron-centered organometallic species are used as catalysts. The Knölker complex, for example, is a transfer hydrogenation catalyst for ketones.[80]
Etymology

As iron has been in use for such a long time, it has many different names in different languages. The source of its chemical symbol Fe is the Latin word ferrum, and its descendants are the names of the element in the Romance languages (for example, French fer, Spanish hierro, and Italian and Portuguese ferro).[81] The word ferrum itself possibly comes from the Semitic languages, via Etruscan, from a root that also gave rise to Old English bræs "brass".[82] The English word iron derives ultimately from Proto-Germanic *isarnan, which is also the source of the German name Eisen. It was most likely borrowed from Celtic *isarnon, which ultimately comes from Proto-Indo-European *is-(e)ro- "powerful, holy" and finally *eis "strong", referencing iron's strength as a metal.[83] Kluge relates *isarnon to Illyric and Latin ira, 'wrath'). The Balto-Slavic names for iron (for example, Russian железо [zhelezo], Polish żelazo, Lithuanian geležis) are the only ones to come directly from the Proto-Indo-European *ghelgh- "iron".[84] In many of these languages, the word for iron may also be used to denote other objects made of iron or steel, or figuratively because of the hardness and strength of the metal.[85] The Chinese tiě (traditional 鐵; simplified 铁) derives from Proto-Sino-Tibetan *hliek,[86] and was borrowed into Japanese as 鉄 tetsu, which also has the native reading kurogane "black metal" (similar to how iron is referenced in the English word blacksmith).[87]
History
Wrought iron

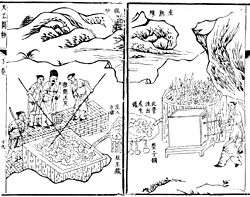
Iron is one of the elements undoubtedly known to the ancient world.[88] It has been worked, or wrought, for millennia. However, iron objects of great age are much rarer than objects made of gold or silver due to the ease with which iron corrodes.[89]

Beads made from meteoric iron in 3500 BC or earlier were found in Gerzah, Egypt by G. A. Wainwright.[90] The beads contain 7.5% nickel, which is a signature of meteoric origin since iron found in the Earth's crust generally has only minuscule nickel impurities. Meteoric iron was highly regarded due to its origin in the heavens and was often used to forge weapons and tools.[90] For example, a dagger made of meteoric iron was found in the tomb of Tutankhamun, containing similar proportions of iron, cobalt, and nickel to a meteorite discovered in the area, deposited by an ancient meteor shower.[91][92][93] Items that were likely made of iron by Egyptians date from 3000 to 2500 BC.[89] Meteoritic iron is comparably soft and ductile and easily forged by cold working but may get brittle when heated because of the nickel content.[94]
The first iron production started in the Middle Bronze Age but it took several centuries before iron displaced bronze. Samples of smelted iron from Asmar, Mesopotamia and Tall Chagar Bazaar in northern Syria were made sometime between 3000 and 2700 BC.[95] The Hittites established an empire in north-central Anatolia around 1600 BC. They appear to be the first to understand the production of iron from its ores and regard it highly in their society.[96] The Hittites began to smelt iron between 1500 and 1200 BC and the practice spread to the rest of the Near East after their empire fell in 1180 BC.[95] The subsequent period is called the Iron Age.
Artifacts of smelted iron are found in India dating from 1800 to 1200 BC,[97] and in the Levant from about 1500 BC (suggesting smelting in Anatolia or the Caucasus).[98][99] Alleged references (compare history of metallurgy in South Asia) to iron in the Indian Vedas have been used for claims of a very early usage of iron in India respectively to date the texts as such. The rigveda term ayas (metal) probably refers to copper and bronze, while iron or śyāma ayas, literally "black metal", first is mentioned in the post-rigvedic Atharvaveda.[100]
Some archaeological evidence suggests iron was smelted in Zimbabwe and southeast Africa as early as the eighth century BC.[101] Iron working was introduced to Greece in the late 11th century BC, from which it spread quickly throughout Europe.[102]
The spread of ironworking in Central and Western Europe is associated with Celtic expansion. According to Pliny the Elder, iron use was common in the Roman era.[90] The annual iron output of the Roman Empire is estimated at 84750 t,[103] while the similarly populous and contemporary Han China produced around 5000 t.[104] In China, iron only appears circa 700–500 BC.[105] Iron smelting may have been introduced into China through Central Asia.[106] The earliest evidence of the use of a blast furnace in China dates to the 1st century AD,[107] and cupola furnaces were used as early as the Warring States period (403–221 BC).[108] Usage of the blast and cupola furnace remained widespread during the Song and Tang Dynasties.[109]
During the Industrial Revolution in Britain, Henry Cort began refining iron from pig iron to wrought iron (or bar iron) using innovative production systems. In 1783 he patented the puddling process for refining iron ore. It was later improved by others, including Joseph Hall.[110]
Cast iron
Cast iron was first produced in China during 5th century BC,[111] but was hardly in Europe until the medieval period.[112][113] The earliest cast iron artifacts were discovered by archaeologists in what is now modern Luhe County, Jiangsu in China. Cast iron was used in ancient China for warfare, agriculture, and architecture.[114] During the medieval period, means were found in Europe of producing wrought iron from cast iron (in this context known as pig iron) using finery forges. For all these processes, charcoal was required as fuel.[115]

Medieval blast furnaces were about 10 feet (3.0 m) tall and made of fireproof brick; forced air was usually provided by hand-operated bellows.[113] Modern blast furnaces have grown much bigger, with hearths fourteen meters in diameter that allow them to produce thousands of tons of iron each day, but essentially operate in much the same way as they did during medieval times.[115]
In 1709, Abraham Darby I established a coke-fired blast furnace to produce cast iron, replacing charcoal, although continuing to use blast furnaces. The ensuing availability of inexpensive iron was one of the factors leading to the Industrial Revolution. Toward the end of the 18th century, cast iron began to replace wrought iron for certain purposes, because it was cheaper. Carbon content in iron was not implicated as the reason for the differences in properties of wrought iron, cast iron, and steel until the 18th century.[95]
Since iron was becoming cheaper and more plentiful, it also became a major structural material following the building of the innovative first iron bridge in 1778. This bridge still stands today as a monument to the role iron played in the Industrial Revolution. Following this, iron was used in rails, boats, ships, aqueducts, and buildings, as well as in iron cylinders in steam engines.[115] Railways have been central to the formation of modernity and ideas of progress[116] and various languages (e.g. French, Spanish, Italian and German) refer to railways as iron road.
Steel
Steel (with smaller carbon content than pig iron but more than wrought iron) was first produced in antiquity by using a bloomery. Blacksmiths in Luristan in western Persia were making good steel by 1000 BC.[95] Then improved versions, Wootz steel by India and Damascus steel were developed around 300 BC and AD 500 respectively. These methods were specialized, and so steel did not become a major commodity until the 1850s.[117]
New methods of producing it by carburizing bars of iron in the cementation process were devised in the 17th century. In the Industrial Revolution, new methods of producing bar iron without charcoal were devised and these were later applied to produce steel. In the late 1850s, Henry Bessemer invented a new steelmaking process, involving blowing air through molten pig iron, to produce mild steel. This made steel much more economical, thereby leading to wrought iron no longer being produced in large quantities.[118]
Foundations of modern chemistry
In 1774, Antoine Lavoisier used the reaction of water steam with metallic iron inside an incandescent iron tube to produce hydrogen in his experiments leading to the demonstration of the conservation of mass, which was instrumental in changing chemistry from a qualitative science to a quantitative one.[119]
Symbolic role
.jpg)
Iron plays a certain role in mythology and has found various usage as a metaphor and in folklore. The Greek poet Hesiod's Works and Days (lines 109–201) lists different ages of man named after metals like gold, silver, bronze and iron to account for successive ages of humanity.[120] The Iron Age was closely related with Rome, and in Ovid's Metamorphoses
The Virtues, in despair, quit the earth; and the depravity of man becomes universal and complete. Hard steel succeeded then.
— Ovid, Metamorphoses, Book I, Iron age, line 160 ff
An example of the importance of iron's symbolic role may be found in the German Campaign of 1813. Frederick William III commissioned then the first Iron Cross as military decoration. Berlin iron jewellery reached its peak production between 1813 and 1815, when the Prussian royal family urged citizens to donate gold and silver jewellery for military funding. The inscription Gold gab ich für Eisen (I gave gold for iron) was used as well in later war efforts.[121]
Production of metallic iron

Industrial routes
The production of iron or steel is a process consisting of two main stages. In the first stage pig iron is produced in a blast furnace. Alternatively, it may be directly reduced. In the second stage, pig iron is converted to wrought iron, steel, or cast iron.[122]

For a few limited purposes when it is needed, pure iron is produced in the laboratory in small quantities by reducing the pure oxide or hydroxide with hydrogen, or forming iron pentacarbonyl and heating it to 250 °C so that it decomposes to form pure iron powder.[42] Another method is electrolysis of ferrous chloride onto an iron cathode.[123]
Blast furnace processing
Industrial iron production starts with iron ores, principally hematite, which has a nominal formula Fe2O3, and magnetite, with the formula Fe3O4. These ores are reduced to the metal in a carbothermic reaction, i.e. by treatment with carbon. The conversion is typically conducted in a blast furnace at temperatures of about 2000 °C. Carbon is provided in the form of coke. The process also contains a flux such as limestone, which is used to remove silicaceous minerals in the ore, which would otherwise clog the furnace. The coke and limestone are fed into the top of the furnace, while a massive blast of air heated to 900 °C, about 4 tons per ton of iron,[113] is forced into the furnace at the bottom.[122]
In the furnace, the coke reacts with oxygen in the air blast to produce carbon monoxide:[122]
- 2 C + O2 → 2 CO
The carbon monoxide reduces the iron ore (in the chemical equation below, hematite) to molten iron, becoming carbon dioxide in the process:[122]
- Fe2O3 + 3 CO → 2 Fe + 3 CO2
Some iron in the high-temperature lower region of the furnace reacts directly with the coke:[122]
- 2 Fe2O3 + 3 C → 4 Fe + 3 CO2
The flux present to melt impurities in the ore is principally limestone (calcium carbonate) and dolomite (calcium-magnesium carbonate). Other specialized fluxes are used depending on the details of the ore. In the heat of the furnace the limestone flux decomposes to calcium oxide (also known as quicklime):[122]
- CaCO3 → CaO + CO2
Then calcium oxide combines with silicon dioxide to form a liquid slag.[122]
- CaO + SiO2 → CaSiO3
The slag melts in the heat of the furnace. In the bottom of the furnace, the molten slag floats on top of the denser molten iron, and apertures in the side of the furnace are opened to run off the iron and the slag separately. The iron, once cooled, is called pig iron, while the slag can be used as a material in road construction or to improve mineral-poor soils for agriculture.[113]

Direct iron reduction
Owing to environmental concerns, alternative methods of processing iron have been developed. "Direct iron reduction" reduces iron ore to a ferrous lump called "sponge" iron or "direct" iron that is suitable for steelmaking.[113] Two main reactions comprise the direct reduction process:
Natural gas is partially oxidized (with heat and a catalyst):[113]
- 2 CH4 + O2 → 2 CO + 4 H2
These gases are then treated with iron ore in a furnace, producing solid sponge iron:[113]
- Fe2O3 + CO + 2 H2 → 2 Fe + CO2 + 2 H2O
Silica is removed by adding a limestone flux as described above.[113]
Thermite
Iron is a byproduct of burning a mixture of aluminum powder and rust powder.
- Fe2O3 + 2 Al → 2 Fe + Al2O3
Further processes

Pig iron is not pure iron, but has 4–5% carbon dissolved in it with small amounts of other impurities like sulfur, magnesium, phosphorus and manganese. As the carbon is the major impurity, the iron (pig iron) becomes brittle and hard.[122] Removing the other impurities results in cast iron, which is used to cast articles in foundries such as stoves, pipes, radiators, lamp-posts and rails.[122]
Alternatively pig iron may be made into steel (with up to about 2% carbon) or wrought iron (commercially pure iron). Various processes have been used for this, including finery forges, puddling furnaces, Bessemer converters, open hearth furnaces, basic oxygen furnaces, and electric arc furnaces. In all cases, the objective is to oxidize some or all of the carbon, together with other impurities. On the other hand, other metals may be added to make alloy steels.[115]
Annealing involves the heating of a piece of steel to 700–800 °C for several hours and then gradual cooling. It makes the steel softer and more workable.[124]
Applications
Metallurgical
Iron is the most widely used of all the metals, accounting for over 90% of worldwide metal production. Its low cost and high strength make it indispensable in engineering applications such as the construction of machinery and machine tools, automobiles, the hulls of large ships, and structural components for buildings. Since pure iron is quite soft, it is most commonly combined with alloying elements to make steel.[125]
α-Iron is a fairly soft metal that can dissolve only a small concentration of carbon (no more than 0.021% by mass at 910 °C).[126] Austenite (γ-iron) is similarly soft and metallic but can dissolve considerably more carbon (as much as 2.04% by mass at 1146 °C). This form of iron is used in the type of stainless steel used for making cutlery, and hospital and food-service equipment.[14]
Commercially available iron is classified based on purity and the abundance of additives. Pig iron has 3.5–4.5% carbon[127] and contains varying amounts of contaminants such as sulfur, silicon and phosphorus. Pig iron is not a saleable product, but rather an intermediate step in the production of cast iron and steel. The reduction of contaminants in pig iron that negatively affect material properties, such as sulfur and phosphorus, yields cast iron containing 2–4% carbon, 1–6% silicon, and small amounts of manganese.[122] Pig iron has a melting point in the range of 1420–1470 K, which is lower than either of its two main components, and makes it the first product to be melted when carbon and iron are heated together.[15] Its mechanical properties vary greatly and depend on the form the carbon takes in the alloy.[11]
"White" cast irons contain their carbon in the form of cementite, or iron carbide (Fe3C).[11] This hard, brittle compound dominates the mechanical properties of white cast irons, rendering them hard, but unresistant to shock. The broken surface of a white cast iron is full of fine facets of the broken iron-carbide, a very pale, silvery, shiny material, hence the appellation. Cooling a mixture of iron with 0.8% carbon slowly below 723 °C to room temperature results in separate, alternating layers of cementite and α-iron, which is soft and malleable and is called pearlite for its appearance. Rapid cooling, on the other hand, does not allow time for this separation and creates hard and brittle martensite. The steel can then be tempered by reheating to a temperature in between, changing the proportions of pearlite and martensite. The end product below 0.8% carbon content is a pearlite-αFe mixture, and that above 0.8% carbon content is a pearlite-cementite mixture.[11]
In gray iron the carbon exists as separate, fine flakes of graphite, and also renders the material brittle due to the sharp edged flakes of graphite that produce stress concentration sites within the material.[128] A newer variant of gray iron, referred to as ductile iron is specially treated with trace amounts of magnesium to alter the shape of graphite to spheroids, or nodules, reducing the stress concentrations and vastly increasing the toughness and strength of the material.[128]
Wrought iron contains less than 0.25% carbon but large amounts of slag that give it a fibrous characteristic.[127] It is a tough, malleable product, but not as fusible as pig iron. If honed to an edge, it loses it quickly. Wrought iron is characterized by the presence of fine fibers of slag entrapped within the metal. Wrought iron is more corrosion resistant than steel. It has been almost completely replaced by mild steel for traditional "wrought iron" products and blacksmithing.
| Country | Iron ore | Pig iron | Direct iron | Steel |
|---|---|---|---|---|
| China | 1,114.9 | 549.4 | 573.6 | |
| Australia | 393.9 | 4.4 | 5.2 | |
| Brazil | 305.0 | 25.1 | 0.011 | 26.5 |
| Japan | 66.9 | 87.5 | ||
| India | 257.4 | 38.2 | 23.4 | 63.5 |
| Russia | 92.1 | 43.9 | 4.7 | 60.0 |
| Ukraine | 65.8 | 25.7 | 29.9 | |
| South Korea | 0.1 | 27.3 | 48.6 | |
| Germany | 0.4 | 20.1 | 0.38 | 32.7 |
| World | 1,594.9 | 914.0 | 64.5 | 1,232.4 |
Mild steel corrodes more readily than wrought iron, but is cheaper and more widely available. Carbon steel contains 2.0% carbon or less,[130] with small amounts of manganese, sulfur, phosphorus, and silicon. Alloy steels contain varying amounts of carbon as well as other metals, such as chromium, vanadium, molybdenum, nickel, tungsten, etc. Their alloy content raises their cost, and so they are usually only employed for specialist uses. One common alloy steel, though, is stainless steel. Recent developments in ferrous metallurgy have produced a growing range of microalloyed steels, also termed 'HSLA' or high-strength, low alloy steels, containing tiny additions to produce high strengths and often spectacular toughness at minimal cost.[130][131][132]
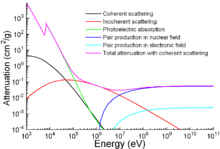
Apart from traditional applications, iron is also used for protection from ionizing radiation. Although it is lighter than another traditional protection material, lead, it is much stronger mechanically. The attenuation of radiation as a function of energy is shown in the graph.[133]
The main disadvantage of iron and steel is that pure iron, and most of its alloys, suffer badly from rust if not protected in some way, a cost amounting to over 1% of the world's economy.[134] Painting, galvanization, passivation, plastic coating and bluing are all used to protect iron from rust by excluding water and oxygen or by cathodic protection. The mechanism of the rusting of iron is as follows:[134]
- Cathode: 3 O2 + 6 H2O + 12 e− → 12 OH−
- Anode: 4 Fe → 4 Fe2+ + 8 e−; 4 Fe2+ → 4 Fe3+ + 4 e−
- Overall: 4 Fe + 3 O2 + 6 H2O → 4 Fe3+ + 12 OH− → 4 Fe(OH)3 or 4 FeO(OH) + 4 H2O
The electrolyte is usually iron(II) sulfate in urban areas (formed when atmospheric sulfur dioxide attacks iron), and salt particles in the atmosphere in seaside areas.[134]
Iron compounds
Although the dominant use of iron is in metallurgy, iron compounds are also pervasive in industry. Iron catalysts are traditionally used in the Haber-Bosch process for the production of ammonia and the Fischer-Tropsch process for conversion of carbon monoxide to hydrocarbons for fuels and lubricants.[135] Powdered iron in an acidic solvent was used in the Bechamp reduction the reduction of nitrobenzene to aniline.[136]
Iron(III) chloride finds use in water purification and sewage treatment, in the dyeing of cloth, as a coloring agent in paints, as an additive in animal feed, and as an etchant for copper in the manufacture of printed circuit boards.[137] It can also be dissolved in alcohol to form tincture of iron, which is used as a medicine to stop bleeding in canaries.[138]
Iron(II) sulfate is used as a precursor to other iron compounds. It is also used to reduce chromate in cement. It is used to fortify foods and treat iron deficiency anemia. Iron(III) sulfate is used in settling minute sewage particles in tank water. Iron(II) chloride is used as a reducing flocculating agent, in the formation of iron complexes and magnetic iron oxides, and as a reducing agent in organic synthesis.[137]
Biological and pathological role
Iron is required for life.[5][139][140] The iron–sulfur clusters are pervasive and include nitrogenase, the enzymes responsible for biological nitrogen fixation. Iron-containing proteins participate in transport, storage and used of oxygen.[5] Iron proteins are involved in electron transfer.[141]
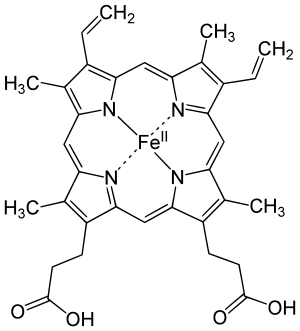
Examples of iron-containing proteins in higher organisms include hemoglobin, cytochrome (see high-valent iron), and catalase.[5][142] The average adult human contains about 0.005% body weight of iron, or about four grams, of which three quarters is in hemoglobin – a level that remains constant despite only about one milligram of iron being absorbed each day,[141] because the human body recycles its hemoglobin for the iron content.[143]
Biochemistry
Iron acquisition poses a problem for aerobic organisms because ferric iron is poorly soluble near neutral pH. Thus, these organisms have developed means to absorb iron as complexes, sometimes taking up ferrous iron before oxidising it back to ferric iron.[5] In particular, bacteria have evolved very high-affinity sequestering agents called siderophores.[144][145][146]
After uptake in human cells, iron storage is precisely regulated.[5][147] A major component of this regulation is the protein transferrin, which binds iron ions absorbed from the duodenum and carries it in the blood to cells.[5][148] Transferrin contains Fe3+ in the middle of a distorted octahedron, bonded to one nitrogen, three oxygens and a chelating carbonate anion that traps the Fe3+ ion: it has such a high stability constant that it is very effective at taking up Fe3+ ions even from the most stable complexes. At the bone marrow, transferrin is reduced from Fe3+ and Fe2+ and stored as ferritin to be incorporated into hemoglobin.[141]
The most commonly known and studied bioinorganic iron compounds (biological iron molecules) are the heme proteins: examples are hemoglobin, myoglobin, and cytochrome P450.[5] These compounds participate in transporting gases, building enzymes, and transferring electrons.[141] Metalloproteins are a group of proteins with metal ion cofactors. Some examples of iron metalloproteins are ferritin and rubredoxin.[141] Many enzymes vital to life contain iron, such as catalase,[149] lipoxygenases,[150] and IRE-BP.[151]
Hemoglobin is an oxygen carrier that occurs in red blood cells and contributes their color, transporting oxygen in the arteries from the lungs to the muscles where it is transferred to myoglobin, which stores it until it is needed for the metabolic oxidation of glucose, generating energy.[5] Here the hemoglobin binds to carbon dioxide, produced when glucose is oxidized, which is transported through the veins by hemoglobin (predominantly as bicarbonate anions) back to the lungs where it is exhaled.[141] In hemoglobin, the iron is in one of four heme groups and has six possible coordination sites; four are occupied by nitrogen atoms in a porphyrin ring, the fifth by an imidazole nitrogen in a histidine residue of one of the protein chains attached to the heme group, and the sixth is reserved for the oxygen molecule it can reversibly bind to.[141] When hemoglobin is not attached to oxygen (and is then called deoxyhemoglobin), the Fe2+ ion at the center of the heme group (in the hydrophobic protein interior) is in a high-spin configuration. It is thus too large to fit inside the porphyrin ring, which bends instead into a dome with the Fe2+ ion about 55 picometers above it. In this configuration, the sixth coordination site reserved for the oxygen is blocked by another histidine residue.[141]
When deoxyhemoglobin picks up an oxygen molecule, this histidine residue moves away and returns once the oxygen is securely attached to form a hydrogen bond with it. This results in the Fe2+ ion switching to a low-spin configuration, resulting in a 20% decrease in ionic radius so that now it can fit into the porphyrin ring, which becomes planar.[141] (Additionally, this hydrogen bonding results in the tilting of the oxygen molecule, resulting in a Fe–O–O bond angle of around 120° that avoids the formation of Fe–O–Fe or Fe–O2–Fe bridges that would lead to electron transfer, the oxidation of Fe2+ to Fe3+, and the destruction of hemoglobin.) This results in a movement of all the protein chains that leads to the other subunits of hemoglobin changing shape to a form with larger oxygen affinity. Thus, when deoxyhemoglobin takes up oxygen, its affinity for more oxygen increases, and vice versa.[141] Myoglobin, on the other hand, contains only one heme group and hence this cooperative effect cannot occur. Thus, while hemoglobin is almost saturated with oxygen in the high partial pressures of oxygen found in the lungs, its affinity for oxygen is much lower than that of myoglobin, which oxygenates even at low partial pressures of oxygen found in muscle tissue.[141] As described by the Bohr effect (named after Christian Bohr, the father of Niels Bohr), the oxygen affinity of hemoglobin diminishes in the presence of carbon dioxide.[141]
Carbon monoxide and phosphorus trifluoride are poisonous to humans because they bind to hemoglobin similarly to oxygen, but with much more strength, so that oxygen can no longer be transported throughout the body. Hemoglobin bound to carbon monoxide is known as carboxyhemoglobin. This effect also plays a minor role in the toxicity of cyanide, but there the major effect is by far its interference with the proper functioning of the electron transport protein cytochrome a.[141] The cytochrome proteins also involve heme groups and are involved in the metabolic oxidation of glucose by oxygen. The sixth coordination site is then occupied by either another imidazole nitrogen or a methionine sulfur, so that these proteins are largely inert to oxygen – with the exception of cytochrome a, which bonds directly to oxygen and thus is very easily poisoned by cyanide.[141] Here, the electron transfer takes place as the iron remains in low spin but changes between the +2 and +3 oxidation states. Since the reduction potential of each step is slightly greater than the previous one, the energy is released step-by-step and can thus be stored in adenosine triphosphate. Cytochrome a is slightly distinct, as it occurs at the mitochondrial membrane, binds directly to oxygen, and transports protons as well as electrons, as follows:[141]
- 4 Cytc2+ + O2 + 8H+
inside → 4 Cytc3+ + 2 H2O + 4H+
outside
Although the heme proteins are the most important class of iron-containing proteins, the iron-sulfur proteins are also very important, being involved in electron transfer, which is possible since iron can exist stably in either the +2 or +3 oxidation states. These have one, two, four, or eight iron atoms that are each approximately tetrahedrally coordinated to four sulfur atoms; because of this tetrahedral coordination, they always have high-spin iron. The simplest of such compounds is rubredoxin, which has only one iron atom coordinated to four sulfur atoms from cysteine residues in the surrounding peptide chains. Another important class of iron-sulfur proteins is the ferredoxins, which have multiple iron atoms. Transferrin does not belong to either of these classes.[141]
The ability of sea mussels to maintain their grip on rocks in the ocean is facilitated by their use of organometallic iron-based bonds in their protein-rich cuticles. Based on synthetic replicas, the presence of iron in these structures increased elastic modulus 770 times, tensile strength 58 times, and toughness 92 times. The amount of stress required to permanently damage them increased 76 times.[153]
Health and diet
Iron is pervasive, but particularly rich sources of dietary iron include red meat, oysters, lentils, beans, poultry, fish, leaf vegetables, watercress, tofu, chickpeas, black-eyed peas, and blackstrap molasses.[5] Bread and breakfast cereals are sometimes specifically fortified with iron.[5][154]
Iron provided by dietary supplements is often found as iron(II) fumarate, although iron(II) sulfate is cheaper and is absorbed equally well.[137] Elemental iron, or reduced iron, despite being absorbed at only one-third to two-thirds the efficiency (relative to iron sulfate),[155] is often added to foods such as breakfast cereals or enriched wheat flour. Iron is most available to the body when chelated to amino acids[156] and is also available for use as a common iron supplement. Glycine, the cheapest and most common amino acid is most often used to produce iron glycinate supplements.[157]
Dietary recommendations
The U.S. Institute of Medicine (IOM) updated Estimated Average Requirements (EARs) and Recommended Dietary Allowances (RDAs) for iron in 2001.[5] The current EAR for iron for women ages 14–18 is 7.9 mg/day, 8.1 for ages 19–50 and 5.0 thereafter (post menopause). For men the EAR is 6.0 mg/day for ages 19 and up. The RDA is 15.0 mg/day for women ages 15–18, 18.0 for 19–50 and 8.0 thereafter. For men, 8.0 mg/day for ages 19 and up. RDAs are higher than EARs so as to identify amounts that will cover people with higher than average requirements. RDA for pregnancy is 27 mg/day and, for lactation, 9 mg/day.[5] For children ages 1–3 years 7 mg/day, 10 for ages 4–8 and 8 for ages 9–13. As for safety, the IOM also sets Tolerable upper intake levels (ULs) for vitamins and minerals when evidence is sufficient. In the case of iron the UL is set at 45 mg/day. Collectively the EARs, RDAs and ULs are referred to as Dietary Reference Intakes.[158]
The European Food Safety Authority (EFSA) refers to the collective set of information as Dietary Reference Values, with Population Reference Intake (PRI) instead of RDA, and Average Requirement instead of EAR. AI and UL defined the same as in United States. For women the PRI is 13 mg/day ages 15–17 years, 16 mg/day for women ages 18 and up who are premenopausal and 11 mg/day postmenopausal. For pregnancy and lactation, 16 mg/day. For men the PRI is 11 mg/day ages 15 and older. For children ages 1 to 14 the PRI increases from 7 to 11 mg/day. The PRIs are higher than the U.S. RDAs, with the exception of pregnancy.[159] The EFSA reviewed the same safety question did not establish a UL.[160]
Infants may require iron supplements if they are bottle-fed cow's milk.[161] Frequent blood donors are at risk of low iron levels and are often advised to supplement their iron intake.[162]
For U.S. food and dietary supplement labeling purposes the amount in a serving is expressed as a percent of Daily Value (%DV). For iron labeling purposes 100% of the Daily Value was 18 mg, and as of May 27, 2016 remained unchanged at 18 mg.[163] A table of all of the old and new adult Daily Values is provided at Reference Daily Intake. The original deadline to be in compliance was July 28, 2018, but on September 29, 2017 the U.S. Food and Drug Administration released a proposed rule that extended the deadline to January 1, 2020 for large companies and January 1, 2021 for small companies.[164]
Deficiency
Iron deficiency is the most common nutritional deficiency in the world.[5][165][166][167] When loss of iron is not adequately compensated by adequate dietary iron intake, a state of latent iron deficiency occurs, which over time leads to iron-deficiency anemia if left untreated, which is characterised by an insufficient number of red blood cells and an insufficient amount of hemoglobin.[168] Children, pre-menopausal women (women of child-bearing age), and people with poor diet are most susceptible to the disease. Most cases of iron-deficiency anemia are mild, but if not treated can cause problems like fast or irregular heartbeat, complications during pregnancy, and delayed growth in infants and children.[169]
Excess
Iron uptake is tightly regulated by the human body, which has no regulated physiological means of excreting iron. Only small amounts of iron are lost daily due to mucosal and skin epithelial cell sloughing, so control of iron levels is primarily accomplished by regulating uptake.[170] Regulation of iron uptake is impaired in some people as a result of a genetic defect that maps to the HLA-H gene region on chromosome 6 and leads to abnormally low levels of hepcidin, a key regulator of the entry of iron into the circulatory system in mammals.[171] In these people, excessive iron intake can result in iron overload disorders, known medically as hemochromatosis.[5] Many people have an undiagnosed genetic susceptibility to iron overload, and are not aware of a family history of the problem. For this reason, people should not take iron supplements unless they suffer from iron deficiency and have consulted a doctor. Hemochromatosis is estimated to be the cause of 0.3 to 0.8% of all metabolic diseases of Caucasians.[172]
Overdoses of ingested iron can cause excessive levels of free iron in the blood. High blood levels of free ferrous iron react with peroxides to produce highly reactive free radicals that can damage DNA, proteins, lipids, and other cellular components. Iron toxicity occurs when the cell contains free iron, which generally occurs when iron levels exceed the availability of transferrin to bind the iron. Damage to the cells of the gastrointestinal tract can also prevent them from regulating iron absorption, leading to further increases in blood levels. Iron typically damages cells in the heart, liver and elsewhere, causing adverse effects that include coma, metabolic acidosis, shock, liver failure, coagulopathy, adult respiratory distress syndrome, long-term organ damage, and even death.[173] Humans experience iron toxicity when the iron exceeds 20 milligrams for every kilogram of body mass; 60 milligrams per kilogram is considered a lethal dose.[174] Overconsumption of iron, often the result of children eating large quantities of ferrous sulfate tablets intended for adult consumption, is one of the most common toxicological causes of death in children under six.[174] The Dietary Reference Intake (DRI) sets the Tolerable Upper Intake Level (UL) for adults at 45 mg/day. For children under fourteen years old the UL is 40 mg/day.[175]
The medical management of iron toxicity is complicated, and can include use of a specific chelating agent called deferoxamine to bind and expel excess iron from the body.[173][176][177]
Cancer
The role of iron in cancer defense can be described as a "double-edged sword" because of its pervasive presence in non-pathological processes.[178] People having chemotherapy may develop iron deficiency and anemia, for which intravenous iron therapy is used to restore iron levels.[179] Iron overload, which may occur from high consumption of red meat,[5] may initiate tumor growth and increase susceptibility to cancer onset,[179] particularly for colorectal cancer.[5]
See also
- El Mutún in Bolivia, where 10% of the world's accessible iron ore is located
- Iron nanoparticle
- Iron–platinum nanoparticle
- Iron fertilization – proposed fertilization of oceans to stimulate phytoplankton growth
- Iron-oxidizing bacteria
- List of countries by iron production
- Pelletising – process of creation of iron ore pellets
- Rustproof iron
- Steel
References
- ↑ Meija, J.; et al. (2016). "Atomic weights of the elements 2013 (IUPAC Technical Report)". Pure and Applied Chemistry. 88 (3): 265–91. doi:10.1515/pac-2015-0305.
- ↑ Ram, R. S. & Bernath, P. F. (2003). "Fourier transform emission spectroscopy of the g4Δ-a4Δ system of FeCl" (PDF). Journal of Molecular Spectroscopy. 221 (2): 261. Bibcode:2003JMoSp.221..261R. doi:10.1016/S0022-2852(03)00225-X.
- ↑ Demazeau, G.; Buffat, B.; Pouchard, M.; Hagenmuller, P. (1982). "Recent developments in the field of high oxidation states of transition elements in oxides stabilization of Six-coordinated Iron(V)". Zeitschrift für anorganische und allgemeine Chemie. 491: 60. doi:10.1002/zaac.19824910109.
- ↑ Lu, J.; Jian, J.; Huang, W.; Lin, H.; Li, J; Zhou, M. (2016). "Experimental and theoretical identification of the Fe(VII) oxidation state in FeO4−". Physical Chemistry Chemical Physics. 18: 31125. Bibcode:2016PCCP...1831125L. doi:10.1039/C6CP06753K.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 "Iron". Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, Oregon. April 2016. Retrieved 6 March 2018.
- 1 2 Kohl, Walter H. (1995). Handbook of materials and techniques for vacuum devices. Springer. pp. 164–167. ISBN 1-56396-387-6.
- 1 2 Kuhn, Howard and Medlin, Dana (prepared under the direction of the ASM International Handbook Committee), eds. (2000). ASM Handbook – Mechanical Testing and Evaluation (PDF). 8. ASM International. p. 275. ISBN 0-87170-389-0.
- ↑ "Hardness Conversion Chart". Maryland Metrics. Archived from the original on 18 June 2015. Retrieved 23 May 2010.
- ↑ Takaji, Kusakawa; Toshikatsu, Otani (1964). "Properties of Various Pure Irons: Study on pure iron I". Tetsu-to-Hagane. 50 (1): 42–47.
- ↑ Raghavan, V. (2004). Materials Science and Engineering. PHI Learning Pvt. Ltd. p. 218. ISBN 81-203-2455-2.
- 1 2 3 4 5 6 Greenwood and Earnshaw, pp. 1074–5
- 1 2 Boehler, Reinhard (2000). "High-pressure experiments and the phase diagram of lower mantle and core materials". Review of Geophysics. American Geophysical Union. 38 (2): 221–245. Bibcode:2000RvGeo..38..221B. doi:10.1029/1998RG000053.
- ↑ Steinmetz, Charles (1917). "fig. 42". Theory and Calculation of Electric Circuits. McGraw-Hill.
- 1 2 Bramfitt, B. L.; Benscoter, Arlan O. (2002). "The Iron Carbon Phase Diagram". Metallographer's guide: practice and procedures for irons and steels. ASM International. pp. 24–28. ISBN 978-0-87170-748-2.
- 1 2 3 4 5 6 7 Greenwood and Earnshaw, pp. 1075–9
- ↑ Stixrude, Lars; Wasserman, Evgeny; Cohen, Ronald E. (1997-11-10). "Composition and temperature of Earth's inner core". Journal of Geophysical Research: Solid Earth. 102 (B11): 24729–24739. Bibcode:1997JGR...10224729S. doi:10.1029/97JB02125.
- ↑ Boehler, Reinhard; Ross, M. (2007). "Properties of Rocks and Minerals_High-Pressure Melting". Mineral Physics. Treatise on Geophysics. 2. Elsevier. pp. 527–541. doi:10.1016/B978-044452748-6.00047-X.
- ↑ Greenwood and Earnshaw, p. 1116
- 1 2 Audi, G.; Bersillon, O.; Blachot, J.; Wapstra, A. H. (2003). "The NUBASE evaluation of nuclear and decay properties" (PDF). Nuclear Physics A. 729: 3–128. Bibcode:2003NuPhA.729....3A. doi:10.1016/j.nuclphysa.2003.11.001. Archived from the original (PDF) on 2008-09-23.
- ↑ Rugel, G.; Faestermann, T.; Knie, K.; Korschinek, G.; Poutivtsev, M.; Schumann, D.; Kivel, N.; Günther-Leopold, I.; Weinreich, R.; Wohlmuther, M. (2009). "New Measurement of the 60Fe Half-Life". Physical Review Letters. 103 (7): 072502. Bibcode:2009PhRvL.103g2502R. doi:10.1103/PhysRevLett.103.072502. PMID 19792637.
- ↑ Dauphas, N.; Rouxel, O. (2006). "Mass spectrometry and natural variations of iron isotopes" (PDF). Mass Spectrometry Reviews. 25 (4): 515–550. Bibcode:2006MSRv...25..515D. doi:10.1002/mas.20078. PMID 16463281. Archived from the original (PDF) on 10 June 2010.
- ↑ Mostefaoui, S.; Lugmair, G. W.; Hoppe, P.; El Goresy, A. (2004). "Evidence for live 60Fe in meteorites". New Astronomy Reviews. 48: 155–159. Bibcode:2004NewAR..48..155M. doi:10.1016/j.newar.2003.11.022.
- ↑ Fewell, M. P. (1995). "The atomic nuclide with the highest mean binding energy". American Journal of Physics. 63 (7): 653. Bibcode:1995AmJPh..63..653F. doi:10.1119/1.17828.
- 1 2 3 Greenwood and Earnshaw, p. 12
- ↑ Woosley, S.; Janka, T. (2006). "The physics of core collapse supernovae". Nature Physics. 1 (3): 147–154. arXiv:astro-ph/0601261. Bibcode:2005NatPh...1..147W. doi:10.1038/nphys172.
- ↑ McDonald, I.; Sloan, G. C.; Zijlstra, A. A.; Matsunaga, N.; Matsuura, M.; Kraemer, K. E.; Bernard-Salas, J.; Markwick, A. J. (2010). "Rusty Old Stars: A Source of the Missing Interstellar Iron?". The Astrophysical Journal Letters. 717 (2): L92–L97. arXiv:1005.3489. Bibcode:2010ApJ...717L..92M. doi:10.1088/2041-8205/717/2/L92.
- ↑ Bautista, Manuel A.; Pradhan, Anil K. (1995). "Iron and Nickel Abundances in H~II Regions and Supernova Remnants". Bulletin of the American Astronomical Society. 27: 865. Bibcode:1995AAS...186.3707B.
- ↑ Dyson, Freeman J. (1979). "Time without end: Physics and biology in an open universe". Reviews of Modern Physics. 51 (3): 447–460. Bibcode:1979RvMP...51..447D. doi:10.1103/RevModPhys.51.447.
- ↑ Morgan, John W. & Anders, Edward (1980). "Chemical composition of Earth, Venus, and Mercury". Proc. Natl. Acad. Sci. 77 (12): 6973–6977. Bibcode:1980PNAS...77.6973M. doi:10.1073/pnas.77.12.6973. PMC 350422. PMID 16592930.
- ↑ "Pyrrhotite". Mindat.org. Retrieved 2009-07-07.
- ↑ Klein, Cornelis and Cornelius S. Hurlbut, Jr. (1985) Manual of Mineralogy, Wiley, 20th ed, pp. 278–9 ISBN 0-471-80580-7
- ↑ Stark, Anne M. (2007-09-20) Researchers locate mantle’s spin transition zone, leading to clues about earth’s structure. Lawrence Livermore National Laboratory
- ↑ Ferropericlase. Mindat.org
- ↑ Murakami, M.; Ohishi Y.; Hirao N.; Hirose K. (2012). "A perovskitic lower mantle inferred from high-pressure, high-temperature sound velocity data". Nature. 485 (7396): 90–94. Bibcode:2012Natur.485...90M. doi:10.1038/nature11004. PMID 22552097.
- ↑ Sharp, T. (27 November 2014). "Bridgmanite—named at last". Science. 346 (6213): 1057–1058. Bibcode:2014Sci...346.1057S. doi:10.1126/science.1261887. PMID 25430755.
- ↑ Lyons, T. W.; Reinhard, C. T. (2009). "Early Earth: Oxygen for heavy-metal fans". Nature. 461 (7261): 179–181. Bibcode:2009Natur.461..179L. doi:10.1038/461179a. PMID 19741692.
- ↑ Cloud, P. (1973). "Paleoecological Significance of the Banded Iron-Formation". Economic Geology. 68 (7): 1135–1143. doi:10.2113/gsecongeo.68.7.1135.
- ↑ Dickinson, Robert E. (1964). Germany: A regional and economic geography (2nd ed.). London: Methuen.
- ↑ Naturwerksteine in Baden-Württemberg. Landesamt für Geologie, Rohstoffe und Bergbau, Baden-Württemberg
- ↑ "Tales From The Riverbank". Minerva Stone Conservation. Retrieved 22 September 2015.
- ↑ Klingelhöfer, G.; Morris, R. V.; Souza, P. A.; Rodionov, D.; Schröder, C. (2007). "Two earth years of Mössbauer studies of the surface of Mars with MIMOS II". Hyperfine Interactions. 170: 169–177. Bibcode:2006HyInt.170..169K. doi:10.1007/s10751-007-9508-5.
- 1 2 Greenwood and Earnshaw, p. 1071
- ↑ Emiliani, Cesare (1992). "Planet earth: cosmology, geology, and the evolution of life and environment". Cambridge [England] ; New York : Cambridge University Press. Cambridge University Press: 152. Bibcode:1992pecg.book.....E. ISBN 978-0-521-40949-0.
- ↑ Metal Stocks in Society: Scientific synthesis, 2010, International Resource Panel, UNEP
- ↑ Greenwood and Earnshaw, p. 905
- 1 2 Greenwood and Earnshaw, p. 1070
- ↑ Huang, Wei; Xu, Wen-Hua; Schwarz, W. H. E.; Li, Jun (2016-05-02). "On the Highest Oxidation States of Metal Elements in MO4 Molecules (M = Fe, Ru, Os, Hs, Sm, and Pu)". Inorganic Chemistry. 55 (9): 4616–4625. doi:10.1021/acs.inorgchem.6b00442.
- ↑ Lu, Jun-Bo; Jian, Jiwen; Huang, Wei; Lin, Hailu; Li, Jun; Zhou, Mingfei (2016-11-16). "Experimental and theoretical identification of the Fe(VII) oxidation state in FeO4−". Phys. Chem. Chem. Phys. 18 (45): 31125–31131. Bibcode:2016PCCP...1831125L. doi:10.1039/c6cp06753k. PMID 27812577.
- ↑ Nam, Wonwoo (2007). "High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions" (PDF). Accounts of Chemical Research. 40 (7): 522–531. doi:10.1021/ar700027f. PMID 17469792.
- 1 2 3 4 5 6 Holleman, Arnold F.; Wiberg, Egon; Wiberg, Nils (1985). "Iron". Lehrbuch der Anorganischen Chemie (in German) (91–100 ed.). Walter de Gruyter. pp. 1125–1146. ISBN 3-11-007511-3.
- ↑ Reiff, William Michael; Long, Gary J. (1984). "Mössbauer Spectroscopy and the Coordination Chemistry of Iron". Mössbauer spectroscopy applied to inorganic chemistry. Springer. pp. 245–283. ISBN 978-0-306-41647-7.
- ↑ Ware, Mike (1999). "An introduction in monochrome". Cyanotype: the history, science and art of photographic printing in Prussian blue. NMSI Trading Ltd. pp. 11–19. ISBN 978-1-900747-07-3.
- ↑ Gmelin, Leopold (1852). "Mercury and Iron". Hand-book of chemistry. 6. Cavendish Society. pp. 128–129.
- 1 2 3 Greenwood and Earnshaw, p. 1079
- 1 2 3 4 Greenwood and Earnshaw, p. 1082–4
- 1 2 3 4 5 Greenwood and Earnshaw, pp. 1088–91
- 1 2 3 Greenwood and Earnshaw, p. 1091–7
- ↑ Clausen, C. A.; Good, M. L. (1968). "Stabilization of the hexachloroferrate(III) anion by the methylammonium cation". Inorganic Chemistry. 7 (12): 2662–2663. doi:10.1021/ic50070a047.
- ↑ James, B. D.; Bakalova, M.; Lieseganga, J.; Reiff, W. M.; Hockless, D. C. R.; Skelton, B. W.; White, A. H. (1996). "The hexachloroferrate(III) anion stabilized in hydrogen bonded packing arrangements. A comparison of the X-ray crystal structures and low temperature magnetism of tetrakis(methylammonium) hexachloroferrate(III) chloride (I) and tetrakis(hexamethylenediammonium) hexachloroferrate(III) tetrachloroferrate(III) tetrachloride (II)". Inorganica Chimica Acta. 247 (2): 169–174. doi:10.1016/0020-1693(95)04955-X.
- ↑ Giannoccaro, P.; Sacco, A. (1977). "Bis[ethylenebis(diphenylphosphine)]-Hydridoiron Complexes". Inorg. Synth. Inorganic Syntheses. 17: 69–72. doi:10.1002/9780470132487.ch19. ISBN 9780470132487.
- ↑ Lee, J.; Jung, G.; Lee, S. W. (1998). "Structure of trans-chlorohydridobis(diphenylphosphinoethane)iron(II)". Bull. Korean Chem. Soc. 19 (2): 267–269.
- ↑ Echigo, Takuya; Kimata, Mitsuyoshi (2008). "Single-crystal X-ray diffraction and spectroscopic studies on humboldtine and lindbergite: weak Jahn–Teller effect of Fe2+ ion". Phys. Chem. Minerals. 35 (8): 467–475. Bibcode:2008PCM....35..467E. doi:10.1007/s00269-008-0241-7.
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1984). Chemistry of the Elements. Oxford: Pergamon Press. pp. 1282–86. ISBN 0-08-022057-6. .
- ↑ Greenwood and Earnshaw, p. 1104
- ↑ Kealy, T. J.; Pauson, P. L. (1951). "A New Type of Organo-Iron Compound". Nature. 168 (4285): 1039–1040. Bibcode:1951Natur.168.1039K. doi:10.1038/1681039b0.
- 1 2 Laszlo, P.; Hoffmann, R. (2000). "Ferrocene: Ironclad History of Rashomon Tale?" (PDF). Angew. Chem. Int. Ed. 39 (1): 123–124. doi:10.1002/(SICI)1521-3773(20000103)39:1<123::AID-ANIE123>3.0.CO;2-Z. PMID 10649350.
- 1 2 Federman Neto, A.; Pelegrino, A. C.; Darin, V. A. (2004). "Ferrocene: 50 Years of Transition Metal Organometallic Chemistry — From Organic and Inorganic to Supramolecular Chemistry". ChemInform. 35 (43). doi:10.1002/chin.200443242. (Abstract; original published in Trends Organomet. Chem., 4:147–169, 2002)
- ↑ Wilkinson, G.; Rosenblum, M.; Whiting, M. C.; Woodward, R. B. (1952). "The Structure of Iron Bis-Cyclopentadienyl". J. Am. Chem. Soc. 74 (8): 2125–2126. doi:10.1021/ja01128a527.
- ↑ Werner, H. (2008). Landmarks in Organo-Transition Metal Chemistry: A Personal View. New York: Springer Science. pp. 161–163. ISBN 978-0-387-09847-0.
- ↑ Fischer, E. O.; Pfab, W. (1952). "Zur Kristallstruktur der Di-Cyclopentadienyl-Verbindungen des zweiwertigen Eisens, Kobalts und Nickels". Z. Anorg. Allg. Chem. (in German). 7 (6): 377–379. doi:10.1002/zaac.19532740603.
- ↑ Eiland, P. F.; Pepinsky, R. (1952). "X-ray Examination of Iron Biscyclopentadienyl". J. Am. Chem. Soc. 74 (19): 4971. doi:10.1021/ja01139a527.
- ↑ Mingos, D. M. P. (2001). "A Historical Perspective on Dewar's Landmark Contribution to Organometallic Chemistry". J. Organomet. Chem. 635 (1–2): 1–8. doi:10.1016/S0022-328X(01)01155-X.
- ↑ Mehrotra, R. C.; Singh, A. (2007). Organometallic Chemistry: A Unified Approach (2nd ed.). New Delhi: New Age International. pp. 261–267. ISBN 978-81-224-1258-1.
- ↑ Zeise, W. C. (1831). "Von der Wirkung zwischen Platinchlorid und Alkohol, und von den dabei entstehenden neuen Substanzen". Annalen der Physik (in German). 97 (4): 497–541. Bibcode:1831AnP....97..497Z. doi:10.1002/andp.18310970402.
- ↑ Hunt, L. B. (1984). "The First Organometallic Compounds: William Christopher Zeise and his Platinum Complexes" (PDF). Platinum Metals Rev. 28 (2): 76–83.
- ↑ Leigh, G. J.; Winterton, N., eds. (2002). Modern Coordination Chemistry: The Legacy of Joseph Chatt. Cambridge, UK: RSC Publishing. pp. 101–110. ISBN 0-85404-469-8.
- ↑ "The Nobel Prize in Chemistry 1973". Nobel Foundation. Retrieved 12 September 2010.
- ↑ Wilkinson, G. (1956). "Ferrocene". Org. Synth. 36: 31. doi:10.15227/orgsyn.036.0031.
- ↑ Bozak, R. E. (1966). "Acetylation of Ferrocene: A Chromatography Experiment for Elementary Organic Laboratory". J. Chem. Educ. 43 (2): 73. Bibcode:1966JChEd..43...73B. doi:10.1021/ed043p73.
- ↑ Bullock, R. M. (11 September 2007). "An Iron Catalyst for Ketone Hydrogenations under Mild Conditions". Angew. Chem. Int. Ed. 46 (39): 7360–7363. doi:10.1002/anie.200703053.
- ↑ "26 Iron". Elements.vanderkrogt.net. Retrieved 2008-09-12.
- ↑ Harper, Douglas (2001–16). "ferro-". etymonline.com. Retrieved 7 August 2016.
- ↑ Harper, Douglas (2001–16). "iron". etymonline.com. Retrieved 7 August 2016.
- ↑ Gamkrelidze, Thomas V.; Ivanov, Vjaceslav V. (1995). Indo-European and the Indo-Europeans: A Reconstruction and Historical Analysis of a Proto-Language and Proto-Culture. Walter de Gruyter. p. 615. ISBN 978-3-11-081503-0.
- ↑ Charlton T. Lewis; Charles Short (1879). A Latin Dictionary. Oxford: Clarendon Press.
- ↑ Coblin, W. South (1986). A Sinologist's Handlist of Sino-Tibetan Lexical Correspondences. Monumenta Serica Monograph Series. 18. Nettetal: Steyler.
- ↑ 1988, 国語大辞典(新装版) (Kokugo Dai Jiten, Revised Edition) (in Japanese), Tōkyō: Shogakukan
- ↑ Weeks 1968, p. 4.
- 1 2 Weeks 1968, p. 29.
- 1 2 3 Weeks 1968, p. 31.
- ↑ Bjorkman, Judith Kingston (1973). "Meteors and Meteorites in the ancient Near East". Meteoritics. 8 (2): 91–132. doi:10.1111/j.1945-5100.1973.tb00146.x.
- ↑ Comelli, Daniela; d'Orazio, Massimo; Folco, Luigi; El-Halwagy, Mahmud; Frizzi, Tommaso; Alberti, Roberto; Capogrosso, Valentina; Elnaggar, Abdelrazek; Hassan, Hala; Nevin, Austin; Porcelli, Franco; Rashed, Mohamed G; Valentini, Gianluca (2016). "The meteoritic origin of Tutankhamun's iron dagger blade". Meteoritics & Planetary Science. 51 (7): 1301–1309. Bibcode:2016M&PS...51.1301C. doi:10.1111/maps.12664.
- ↑ Walsh, Declan (2 June 2016). "King Tut's Dagger Made of 'Iron From the Sky,' Researchers Say". The New York Times. Retrieved 4 June 2016.
... the blade's composition of iron, nickel and cobalt was an approximate match for a meteorite that landed in northern Egypt. The result "strongly suggests an extraterrestrial origin"
- ↑ Ure, Andrew (1843). Technisches wörterbuch oder Handbuch der Gewerbskunde ... : Bearb. nach Dr. Andrew Ure's Dictionary of arts, manufactures and mines (in German). G. Haase. p. 492.
- 1 2 3 4 Weeks 1968, p. 32.
- ↑ McNutt, Paula (1990 1). The Forging of Israel: Iron Technology, Symbolism and Tradition in Ancient Society. A&C Black.
- ↑ Tewari, Rakesh. "The origins of Iron Working in India: New evidence from the Central Ganga plain and the Eastern Vindhyas" (PDF). State Archaeological Department. Retrieved 23 May 2010.
- ↑ Photos, E. (1989). "The Question of Meteoritic versus Smelted Nickel-Rich Iron: Archaeological Evidence and Experimental Results". World Archaeology. Taylor & Francis, Ltd. 20 (3): 403–421. doi:10.1080/00438243.1989.9980081. JSTOR 124562.
- ↑ Muhly, James D. (2003). "Metalworking/Mining in the Levant". In Lake, Richard Winona. Near Eastern Archaeology IN: Eisenbrauns. 180. pp. 174–183.
- ↑ Witzel, Michael (2001), "Autochthonous Aryans? The Evidence from Old Indian and Iranian Texts", in Electronic Journal of Vedic Studies (EJVS) 7-3, pp. 1–93
- ↑ Weeks, p. 33, quoting Cline, Walter (1937) "Mining and Metallurgy in Negro Africa," George Banta Publishing Co., Menasha, Wis., pp. 17–23.
- ↑ Riederer, Josef; Wartke, Ralf-B. (2009) "Iron", Cancik, Hubert; Schneider, Helmuth (eds.): Brill's New Pauly, Brill.
- ↑ Craddock, Paul T. (2008): "Mining and Metallurgy", in: Oleson, John Peter (ed.): The Oxford Handbook of Engineering and Technology in the Classical World, Oxford University Press, ISBN 978-0-19-518731-1, p. 108
- ↑ Wagner, Donald B. (2001) "The State and the Iron Industry in Han China", NIAS Publishing, Copenhagen, ISBN 87-87062-77-1, p. 73
- ↑ Sawyer, Ralph D. and Sawyer, Mei-chün (1993). The Seven Military Classics of Ancient China. Boulder: Westview. ISBN 0465003044. p. 10.
- ↑ Pigott, Vincent C. (1999). The Archaeometallurgy of the Asian Old World. Philadelphia: University of Pennsylvania Museum of Archaeology and Anthropology. ISBN 0-924171-34-0, p. 8.
- ↑ Golas, Peter J. (1999). Science and Civilisation in China: Volume 5, Chemistry and Chemical Technology, Part 13, Mining. Cambridge University Press. p. 152. ISBN 978-0-521-58000-7.
earliest blast furnace discovered in China from about the first century AD
- ↑ Pigott, Vincent C. (1999). The Archaeometallurgy of the Asian Old World. Philadelphia: University of Pennsylvania Museum of Archaeology and Anthropology. ISBN 0-924171-34-0, p. 191.
- ↑ The Coming of the Ages of Steel. Brill Archive. 1961. p. 54.
- ↑ Mott, R.A (2014). "Dry and Wet Puddling". Transactions of the Newcomen Society. 49: 156–157. doi:10.1179/tns.1977.011.
- ↑ Wagner, Donald B. (2003). "Chinese blast furnaces from the 10th to the 14th century" (PDF). Historical Metallurgy. 37 (1): 25–37. Archived from the original (PDF) on 7 January 2018. Retrieved 7 January 2018. originally published in Wagner, Donald B. (2001). "Chinese blast furnaces from the 10th to the 14th century". West Asian Science, Technology, and Medicine. 18: 41–74.
- ↑ Giannichedda, Enrico (2007): "Metal production in Late Antiquity", in Technology in Transition AD 300–650 Lavan, L.; Zanini, E. and Sarantis, A.(eds.), Brill, Leiden; ISBN 90-04-16549-5, p. 200.
- 1 2 3 4 5 6 7 8 Biddle, Verne; Parker, Gregory. Chemistry, Precision and Design. A Beka Book, Inc.
- ↑ Wagner, Donald B. (1993). Iron and Steel in Ancient China. BRILL. pp. 335–340. ISBN 978-90-04-09632-5.
- 1 2 3 4 Greenwood and Earnshaw, p. 1072
- ↑ Schivelbusch, G. (1986) The Railway Journey: Industrialization and Perception of Time and Space in the 19th Century. Oxford: Berg.
- ↑ Spoerl, Joseph S. A Brief History of Iron and Steel Production Archived 2 June 2010 at the Wayback Machine.. Saint Anselm College
- ↑ Enghag, Per (8 January 2008). Encyclopedia of the Elements: Technical Data – History – Processing – Applications. pp. 190–191. ISBN 978-3-527-61234-5.
- ↑ Whitaker, Robert D (1975). "An historical note on the conservation of mass". Journal of Chemical Education. 52 (10): 658. Bibcode:1975JChEd..52..658W. doi:10.1021/ed052p658.
- ↑ Fontenrose, Joseph (1974). "Work, Justice, and Hesiod's Five Ages". Classical Philology. 69: 1–16. doi:10.1086/366027. JSTOR 268960.
- ↑ Schmidt, Eva (1981) Der preußische Eisenkunstguss. (Art of Prussian cast iron) Technik, Geschichte, Werke, Künstler. Verlag Mann, Berlin, ISBN 3-7861-1130-8
- 1 2 3 4 5 6 7 8 9 10 Greenwood and Earnshaw, p. 1073
- ↑ Lux, H. (1963) "Metallic Iron" in Handbook of Preparative Inorganic Chemistry, 2nd Ed. G. Brauer (ed.), Academic Press, NY. Vol. 2. pp. 1490–1.
- ↑ Verhoeven, J.D. (1975) Fundamentals of Physical Metallurgy, Wiley, New York, p. 326
- ↑ Greenwood and Earnshaw, pp. 1070–1
- ↑ Martin, John Wilson (2007). Concise encyclopedia of the structure of materials. Elsevier. p. 183. ISBN 0-08-045127-6.
- 1 2 Camp, James McIntyre; Francis, Charles Blaine (1920). The Making, Shaping and Treating of Steel. Pittsburgh: Carnegie Steel Company. pp. 173–174. ISBN 1-147-64423-3.
- 1 2 Smith, William F.; Hashemi, Javad (2006), Foundations of Materials Science and Engineering (4th ed.), McGraw-Hill, p. 431, ISBN 0-07-295358-6.
- ↑ Steel Statistical Yearbook 2010. World Steel Association
- 1 2 "Classification of Carbon and Low-Alloy Steels". Retrieved 5 January 2008.
- ↑ HSLA Steel, 2002-11-15, archived from the original on 3 January 2010, retrieved 2008-10-11.
- ↑ Oberg, E.; et al. (1996), "Machinery's Handbook", New York: Industrial Press (25th ed.), Industrial Press Inc: 440–2, Bibcode:1984msh..book.....R
- ↑ Rokni, Sayed H.; Cossairt, J. Donald; Liu, James C. (January 2008). "Radiation Shielding at High-Energy Electron and Proton Accelerators" (PDF). Retrieved 6 August 2016.
- 1 2 3 Greenwood and Earnshaw, p. 1076
- ↑ Kolasinski, Kurt W. (2002). "Where are Heterogenous Reactions Important". Surface science: foundations of catalysis and nanoscience. John Wiley and Sons. pp. 15–16. ISBN 978-0-471-49244-3.
- ↑ McKetta, John J. (1989). "Nitrobenzene and Nitrotoluene". Encyclopedia of Chemical Processing and Design: Volume 31 – Natural Gas Liquids and Natural Gasoline to Offshore Process Piping: High Performance Alloys. CRC Press. pp. 166–167. ISBN 978-0-8247-2481-8.
- 1 2 3 Wildermuth, Egon; Stark, Hans; Friedrich, Gabriele; Ebenhöch, Franz Ludwig; Kühborth, Brigitte; Silver, Jack; Rituper, Rafael (2000). "Iron Compounds". Ullmann's Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.a14_591. ISBN 3-527-30673-0.
- ↑ Stroud, Robert (1933). Diseases of Canaries. Canary Publishers Company. p. 203. ISBN 978-1-4465-4656-7.
- ↑ Dlouhy, Adrienne C.; Outten, Caryn E. (2013). Banci, Lucia, ed. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. doi:10.1007/978-94-007-5561-1_8. ISBN 978-94-007-5560-4. electronic-book ISBN 978-94-007-5561-1
- ↑ Yee, Gereon M.; Tolman, William B. (2015). Peter M.H. Kroneck; Martha E. Sosa Torres, eds. Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases. Metal Ions in Life Sciences. 15. Springer. pp. 131–204. doi:10.1007/978-3-319-12415-5_5.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Greenwood and Earnshaw, pp. 1098–1104
- ↑ Lippard, S. J.; Berg, J. M. (1994). Principles of Bioinorganic Chemistry. Mill Valley: University Science Books. ISBN 0-935702-73-3.
- ↑ Kikuchi, G.; Yoshida, T.; Noguchi, M. (2005). "Heme oxygenase and heme degradation". Biochemical and Biophysical Research Communications. 338 (1): 558–567. doi:10.1016/j.bbrc.2005.08.020. PMID 16115609.
- ↑ Neilands, J. B. (1995). "Siderophores: structure and function of microbial iron transport compounds". The Journal of Biological Chemistry. 270 (45): 26723–6. doi:10.1074/jbc.270.45.26723. PMID 7592901.
- ↑ Neilands, J. B. (1981). "Microbial Iron Compounds". Annual Review of Biochemistry. 50 (1): 715–31. doi:10.1146/annurev.bi.50.070181.003435. PMID 6455965.
- ↑ Boukhalfa, Hakim; Crumbliss, Alvin L. (2002). "Chemical aspects of siderophore mediated iron transport". BioMetals. 15 (4): 325–39. doi:10.1023/A:1020218608266. PMID 12405526.
- ↑ Nanami, M.; Ookawara, T.; Otaki, Y.; Ito, K.; Moriguchi, R.; Miyagawa, K.; Hasuike, Y.; Izumi, M.; Eguchi, H.; Suzuki, K.; Nakanishi, T. (2005). "Tumor necrosis factor-α-induced iron sequestration and oxidative stress in human endothelial cells". Arteriosclerosis, Thrombosis, and Vascular Biology. 25 (12): 2495–2501. doi:10.1161/01.ATV.0000190610.63878.20. PMID 16224057.
- ↑ Rouault, Tracey A. (2003). "How Mammals Acquire and Distribute Iron Needed for Oxygen-Based Metabolism". PLoS Biology. 1 (3): e9. doi:10.1371/journal.pbio.0000079. PMC 300689. PMID 14691550.
- ↑ Boon EM, Downs A, Marcey D. "Proposed Mechanism of Catalase". Catalase: H2O2: H2O2 Oxidoreductase: Catalase Structural Tutorial. Retrieved 2007-02-11.
- ↑ Boyington JC, Gaffney BJ, Amzel LM (1993). "The three-dimensional structure of an arachidonic acid 15-lipoxygenase". Science. 260 (5113): 1482–1486. Bibcode:1993Sci...260.1482B. doi:10.1126/science.8502991. PMID 8502991.
- ↑ Gray, N. K.; Hentze, M. W. (Aug 1994). "Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs". EMBO J. 13 (16): 3882–3891. PMC 395301. PMID 8070415.
- ↑ Gregory B. Vásquez; Xinhua Ji; Clara Fronticelli; Gary L. Gilliland (1998). "Human Carboxyhemoglobin at 2.2 Å Resolution: Structure and Solvent Comparisons of R-State, R2-State and T-State Hemoglobins". Acta Crystallogr. D. 54 (3): 355–366. doi:10.1107/S0907444997012250.
- ↑ Sanderson, K (2017). "Mussels' iron grip inspires strong and stretchy polymer". Chemical & Engineering News. American Chemical Society. 95 (44): 8. doi:10.1021/cen-09544-notw3. Retrieved 2 November 2017.
- ↑ Food Standards Agency – Eat well, be well – Iron deficiency Archived 8 August 2006 at the Wayback Machine.. Eatwell.gov.uk (5 March 2012). Retrieved on 27 June 2012.
- ↑ Hoppe, M.; Hulthén, L.; Hallberg, L. (2005). "The relative bioavailability in humans of elemental iron powders for use in food fortification". European Journal of Nutrition. 45 (1): 37–44. doi:10.1007/s00394-005-0560-0. PMID 15864409.
- ↑ Pineda, O.; Ashmead, H. D. (2001). "Effectiveness of treatment of iron-deficiency anemia in infants and young children with ferrous bis-glycinate chelate". Nutrition. 17 (5): 381–4. doi:10.1016/S0899-9007(01)00519-6. PMID 11377130.
- ↑ Ashmead, H. DeWayne (1989). Conversations on Chelation and Mineral Nutrition. Keats Publishing. ISBN 0-87983-501-X.
- ↑ "Iron". Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Iron (PDF). National Academy Press. 2001. pp. 290–393. ISBN 0-309-07279-4. PMID 25057538.
- ↑ "Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies" (PDF). European Food Safety Authority. 2017.
- ↑ "Tolerable Upper Intake Levels For Vitamins And Minerals" (PDF). European Food Safety Authority. 2006.
- ↑ "Iron Deficiency Anemia". MediResource. Retrieved 17 December 2008.
- ↑ Milman, N. (1996). "Serum ferritin in Danes: studies of iron status from infancy to old age, during blood donation and pregnancy". International Journal of Hematology. 63 (2): 103–35. doi:10.1016/0925-5710(95)00426-2. PMID 8867722.
- ↑ "Federal Register May 27, 2016 Food Labeling: Revision of the Nutrition and Supplement Facts Labels. FR page 33982" (PDF).
- ↑ "Changes to the Nutrition Facts Panel – Compliance Date". U.S. Food and Drug Administration
- ↑ Centers for Disease Control and Prevention (2002). "Iron deficiency – United States, 1999–2000". MMWR. 51 (40): 897–9. PMID 12418542.
- ↑ Hider, Robert C.; Kong, Xiaole (2013). "Chapter 8. Iron: Effect of Overload and Deficiency". In Astrid Sigel, Helmut Sigel and Roland K. O. Sigel. Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. 13. Springer. pp. 229–294. doi:10.1007/978-94-007-7500-8_8.
- ↑ Dlouhy, Adrienne C.; Outten, Caryn E. (2013). "Chapter 8.4 Iron Uptake, Trafficking and Storage". In Banci, Lucia (Ed.). Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. doi:10.1007/978-94-007-5561-1_8. ISBN 978-94-007-5560-4. electronic-book ISBN 978-94-007-5561-1
- ↑ CDC Centers for Disease Control and Prevention (3 April 1998). "Recommendations to Prevent and Control Iron Deficiency in the United States". Morbidity and Mortality Weekly Report (MMWR). 47 (RR3): 1. Retrieved 12 August 2014.
- ↑ Centers for Disease Control and Prevention. "Iron and Iron Deficiency". Retrieved 12 August 2014.
- ↑ Ramzi S. Cotran; Vinay Kumar; Tucker Collins; Stanley Leonard Robbins (1999). Robbins pathologic basis of disease. Saunders. ISBN 978-0-7216-7335-6. Retrieved 27 June 2012.
- ↑ Ganz T (August 2003). "Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation". Blood. 102 (3): 783–8. doi:10.1182/blood-2003-03-0672. PMID 12663437.
- ↑ Durupt, S.; Durieu, I.; Nové-Josserand, R.; Bencharif, L.; Rousset, H.; Vital Durand, D. (2000). "Hereditary hemochromatosis". Rev Med Interne. 21 (11): 961–71. doi:10.1016/S0248-8663(00)00252-6. PMID 11109593.
- 1 2 Cheney, K.; Gumbiner, C.; Benson, B.; Tenenbein, M. (1995). "Survival after a severe iron poisoning treated with intermittent infusions of deferoxamine". J Toxicol Clin Toxicol. 33 (1): 61–6. doi:10.3109/15563659509020217. PMID 7837315.
- 1 2 "Toxicity, Iron". Medscape. Retrieved 23 May 2010.
- ↑ Dietary Reference Intakes (DRIs): Recommended Intakes for Individuals (PDF), Food and Nutrition Board, Institute of Medicine, National Academies, 2004, archived from the original (PDF) on 14 March 2013, retrieved 2009-06-09
- ↑ Tenenbein, M. (1996). "Benefits of parenteral deferoxamine for acute iron poisoning". J Toxicol Clin Toxicol. 34 (5): 485–489. doi:10.3109/15563659609028005. PMID 8800185.
- ↑ Wu H, Wu T, Xu X, Wang J, Wang J (May 2011). "Iron toxicity in mice with collagenase-induced intracerebral hemorrhage". J Cereb Blood Flow Metab. 31 (5): 1243–50. doi:10.1038/jcbfm.2010.209. PMC 3099628. PMID 21102602.
- ↑ Thévenod, Frank (2018). "Chapter 15. Iron and Its Role in Cancer Defense: A Double-Edged Sword". In Sigel, Astrid; Sigel, Helmut; Freisinger, Eva; Sigel, Roland K. O. Metallo-Drugs: Development and Action of Anticancer Agents. 18. Berlin: de Gruyter GmbH. pp. 437–467. doi:10.1515/9783110470734-021.
- 1 2 Beguin, Y; Aapro, M; Ludwig, H; Mizzen, L; Osterborg, A (2014). "Epidemiological and nonclinical studies investigating effects of iron in carcinogenesis--a critical review". Critical Reviews in Oncology/Hematology. 89 (1): 1–15. doi:10.1016/j.critrevonc.2013.10.008. PMID 24275533.
Bibliography
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 0-08-037941-9.
- Weeks, Mary Elvira; Leichester, Henry M. (1968). "Elements Known to the Ancients". Discovery of the Elements. Easton, PA: Journal of Chemical Education. pp. 29–40. ISBN 0-7661-3872-0. LCCN 68-15217.
Further reading
- H. R. Schubert, History of the British Iron and Steel Industry ... to 1775 AD (Routledge, London, 1957)
- R. F. Tylecote, History of Metallurgy (Institute of Materials, London 1992).
- R. F. Tylecote, "Iron in the Industrial Revolution" in J. Day and R. F. Tylecote, The Industrial Revolution in Metals (Institute of Materials 1991), 200–60.
External links
| Look up iron in Wiktionary, the free dictionary. |
| Wikimedia Commons has media related to Iron. |
| Wikisource has the text of the 1905 New International Encyclopedia article Iron. |
- It's Elemental – Iron
- Iron at The Periodic Table of Videos (University of Nottingham)
- Metallurgy for the non-Metallurgist
- Iron by J. B. Calvert