Conserved sequence

Sequences are the amino acids for residues 120-180 of the proteins. Residues that are conserved across all sequences are highlighted in grey. Below each site (i.e., position) of the protein sequence alignment is a key denoting conserved sites (*), sites with conservative replacements (:), sites with semi-conservative replacements (.), and sites with non-conservative replacements ( ).[1]
In evolutionary biology, conserved sequences are similar or identical sequences in nucleic acids (DNA and RNA) or proteins across species (orthologous sequences) or within a genome (paralogous sequences). Conservation indicates that a sequence has been maintained by natural selection.
A highly conserved sequence is one that has remained relatively unchanged far back up the phylogenetic tree, and hence far back in geological time. Examples of highly conserved sequences include the RNA components of ribosomes present in all domains of life, the homeobox sequences widespread amongst Eukaryotes, and the tmRNA in Bacteria. The study of sequence conservation overlaps with the fields of genomics, proteomics, evolutionary biology, phylogenetics, bioinformatics and mathematics.
History
The discovery of the role of DNA in inheritance, and observations by Frederick Sanger of variation between animal insulins in 1949,[2] prompted early molecular biologists to study taxonomy from a molecular perspective.[3][4] Studies in the 1960s used DNA hybridization and protein cross-reactivity techniques to measure similarity between known orthologous proteins, such as hemoglobin[5] and Cytochrome C.[6] In 1965, Émile Zuckerkandl and Linus Pauling introduced the concept of the molecular clock,[7] proposing that steady rates of mutation could be used to estimate the time since two organisms diverged. While initial phylogenies closely matched the fossil record, observations that some genes appeared to evolve at different rates led to the development of theories of molecular evolution.[3][4] Margaret Dayhoff's 1966 comparison of ferrodoxin sequences showed that natural selection would act to conserve and optimise protein sequences essential to life.[8]
Mechanisms
Over many generations, nucleic acid sequences in the genome of an evolutionary lineage can gradually change over time due to random mutations and deletions.[9][10] Sequences may also recombine or be deleted due to chromosomal rearrangements. Conserved sequences are sequences which persist in the genome despite such forces, and have slower rates of mutation than the background mutation rate.[11]
Conservation can occur in coding and non-coding nucleic acid sequences. Highly conserved DNA sequences are thought to have functional value, although the role for many highly conserved non-coding DNA sequences is poorly understood. The extent to which a sequence is conserved can be affected by varying selection pressures, its robustness to mutation, population size and genetic drift. Many functional sequences are also modular, containing regions which may be subject to independent selection pressures, such as protein domains.
Coding sequence
In coding sequences, the nucleic acid and amino acid sequence may be conserved to different extents, as the degeneracy of the genetic code means that synonymous mutations in a coding sequence do not affect the amino acid sequence of its protein product.
Amino acid sequences can be conserved to maintain the structure or function of a protein or domain. Conserved proteins undergo fewer amino amino acid replacements, or are more likely to substitute amino acids with similar biochemical properties. Within a sequence, amino acids that are important for folding, structural stability, or that form a binding site may be more highly conserved.
The nucleic acid sequence of a protein coding gene may also be conserved by other selective pressures. The codon usage bias in some organisms may restrict the types of synonymous mutations in a sequence. Nucleic acid sequences that cause secondary structure in the mRNA of a coding gene may be selected against, as some structures may negatively affect translation, or conserved where the mRNA also acts as a functional non-coding RNA.[12][13]
Non-coding
Non-coding sequences important for gene regulation, such as the binding or recognition sites of ribosomes and transcription factors, may be conserved within a genome. For example, the promoter of a conserved gene or operon may also be conserved. As with proteins, nucleic acids that are important for the structure and function of non-coding RNA (ncRNA) can also be conserved. However, sequence conservation in ncRNAs is generally poor compared to protein-coding sequences, and base pairs that contribute to structure or function are often conserved instead.[14][15]
Identification
Conserved sequences are typically identified by bioinformatics approaches based on sequence alignment. Advances in high-throughput DNA sequencing and protein mass spectrometry has substantially increased the availability of protein sequences and whole genomes for comparison since the early 2000s.
Homology search
Conserved sequences may be identified by homology search, using tools such as BLAST, HMMER and Infernal.[16] Homology search tools may take an individual nucleic acid or protein sequence as input, or use statistical models generated from multiple sequence alignments of known related sequences. Statistical models such as profile-HMMs, and RNA covariance models which also incorporate structural information,[17] can be helpful when searching for more distantly related sequences. Input sequences are then aligned against a database of sequences from related individuals or other species. The resulting alignments are then scored based on the number of matching amino acids or bases, and the number of gaps or deletions generated by the alignment. Acceptable conservative substitutions may be identified using substitution matrices such as PAM and BLOSUM. Highly scoring alignments are assumed to be from homologous sequences. The conservation of a sequence may then be inferred by detection of highly similar homologs over a broad phylogenetic range.
Multiple sequence alignment
Multiple sequence alignments can be used to visualise conserved sequences. The CLUSTAL format includes a plain-text key to annotate conserved columns of the alignment, denoting conserved sequence (*), conservative mutations (:), semi-conservative mutations (.), and non-conservative mutations ( )[19] Sequence logos can also show conserved sequence by representing the proportions of characters at each point in the alignment by height.[18]
Genome alignment
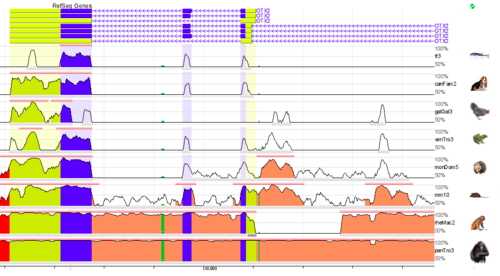
Whole genome alignments (WGAs) may also be used to identify highly conserved regions across species. Currently the accuracy and scalability of WGA tools remains limited due to the computational complexity of dealing with rearrangements, repeat regions and the large size of many eukaryotic genomes.[21] However, WGAs of 30 or more closely related bacteria (prokaryotes) are now increasingly feasible.[22][23]
Scoring systems
Other approaches use measurements of conservation based on statistical tests that attempt to identify sequences which mutate differently to an expected background (neutral) mutation rate.
The GERP (Genomic Evolutionary Rate Profiling) framework scores conservation of genetic sequences across species. This approach estimates the rate of neutral mutation in a set of species from a multiple sequence alignment, and then identifies regions of the sequence that exhibit fewer mutations than expected. These regions are then assigned scores based on the difference between the observed mutation rate and expected background mutation rate. A high GERP score then indicates a highly conserved sequence.[24][25]
Other approaches such as PhyloP and PhyloHMM incorporate statistical phylogenetics methods to compare probability distributions of substitution rates, which allows the detection of both conservation and accelerated mutation. First, a background probability distribution is generated of the number of substitutions expected to occur for a column in a multiple sequence alignment, based on a phylogenetic tree. The estimated evolutionary relationships between the species of interest are used to calculate the significance of any substitutions (i.e. a substitution between two closely related species may be less likely to occur than distantly related ones, and therefore more significant). To detect conservation, a probability distribution is calculated for a subset of the multiple sequence alignment, and compared to the background distribution using a statistical test such as a likelihood-ratio test or score test. P-values generated from comparing the two distributions are then used to identify conserved regions. PhyloHMM uses hidden markov models to generate probability distributions. The PhyloP software package compares probability distributions using a likelihood-ratio test or score test, as well as using a GERP-like scoring system.[26][27][28]
Extreme conservation
Ultra-conserved elements
Ultra-conserved elements or UCEs are sequences that are highly similar or identical across multiple taxonomic groupings. These were first discovered in vertebrates,[29] and have subsequently been identified within widely-differing taxa.[30] While the origin and function of UCEs are poorly understood,[31] they have been used to investigate deep-time divergences in amniotes,[32] insects,[33] and between animals and plants.[34]
Universally conserved genes
The most highly conserved genes are those that can be found in all organisms. These consist mainly of the ncRNAs and proteins required for transcription and translation, which are assumed to have been conserved from the last universal common ancestor of all life.[35]
Genes or gene families that have been found to be universally conserved include GTP-binding elongation factors, Methionine aminopeptidase 2, Serine hydroxymethyltransferase, and ATP transporters.[36] Components of the transcription machinery, such as RNA polymerase and helicases, and of the translation machinery, such as ribosomal RNAs, tRNAs and ribosomal proteins are also universally conserved.[37]
Applications
Phylogenetics and taxonomy
Sets of conserved sequences are often used for generating phylogenetic trees, as it can be assumed that organisms with similar sequences are closely related.[38] The choice of sequences may vary depending on the taxonomic scope of the study. For example, the most highly conserved genes such as the 16S RNA and other ribosomal sequences are useful for reconstructing deep phylogenetic relationships and identifying bacterial phyla in metagenomics studies.[39][40] Sequences that are conserved within a clade but undergo some mutations, such as housekeeping genes, can be used to study species relationships.[41][42][43] The internal transcribed spacer (ITS) region, which is required for spacing conserved rRNA genes but undergoes rapid evolution, is commonly used to classify fungi and strains of rapidly evolving bacteria.[44][45][46][47]
Medical research
As highly conserved sequences often have important biological functions, they can be useful a starting point for identifying the cause of genetic diseases. Many congenital metabolic disorders and Lysosomal storage diseases are the result of changes to individual conserved genes, resulting in missing or faulty enzymes that are the underlying cause of the symptoms of the disease. Genetic diseases may be predicted by identifying sequences that are conserved between humans and lab organisms such as mice[48] or fruit flies,[49] and studying the effects of knock-outs of these genes.[50] Genome-wide association studies can also be used to identify variation in conserved sequences associated with disease or health outcomes.[51][52]
Functional annotation
Identifying conserved sequences can be used to discover and predict functional sequences such as genes.[53] Conserved sequences with a known function, such as protein domains, can also be used to predict the function of a sequence. Databases of conserved protein domains such as Pfam and the Conserved Domain Database can be used to annotate functional domains in predicted protein coding genes.[54]
See also
References
- ↑ "Clustal FAQ #Symbols". Clustal. Retrieved 8 December 2014.
- ↑ Sanger, F. (24 September 1949). "Species Differences in Insulins". Nature. 164 (4169): 529–529. doi:10.1038/164529a0.
- 1 2 Marmur, J; Falkow, S; Mandel, M (October 1963). "New Approaches to Bacterial Taxonomy". Annual Review of Microbiology. 17 (1): 329–372. doi:10.1146/annurev.mi.17.100163.001553.
- 1 2 Pace, N. R.; Sapp, J.; Goldenfeld, N. (17 January 2012). "Phylogeny and beyond: Scientific, historical, and conceptual significance of the first tree of life". Proceedings of the National Academy of Sciences. 109 (4): 1011–1018. doi:10.1073/pnas.1109716109. PMC 3268332.
- ↑ Zuckerlandl, Emile; Pauling, Linus B. (1962). "Molecular disease, evolution, and genetic heterogeneity". Horizons in Biochemistry: 189–225.
- ↑ Margoliash, E (Oct 1963). "PRIMARY STRUCTURE AND EVOLUTION OF CYTOCHROME C". Proc Natl Acad Sci U S A. 50 (4): 672–679. doi:10.1073/pnas.50.4.672. PMC 221244.
- ↑ Zuckerkandl, E; Pauling, LB (1965). "Evolutionary Divergence and Convergence in Proteins". Evolving Genes and Proteins: 96–166. doi:10.1016/B978-1-4832-2734-4.50017-6.
- ↑ Eck, R. V.; Dayhoff, M. O. (15 April 1966). "Evolution of the Structure of Ferredoxin Based on Living Relics of Primitive Amino Acid Sequences". Science. 152 (3720): 363–366. doi:10.1126/science.152.3720.363.
- ↑ Kimura, M (17 February 1968). "Evolutionary Rate at the Molecular Level". Nature. 217 (5129): 624–626. doi:10.1038/217624a0.
- ↑ King, J. L.; Jukes, T. H. (16 May 1969). "Non-Darwinian Evolution". Science. 164 (3881): 788–798. doi:10.1126/science.164.3881.788.
- ↑ Kimura, M; Ohta, T (1974). "On Some Principles Governing Molecular Evolution" (PDF). Proc Natl Acad Sci USA. 71 (7): 2848–2852. doi:10.1073/pnas.71.7.2848. PMC 388569. PMID 4527913.
- ↑ Chamary, JV; Hurst, Laurence D (2005). "Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals". Genome Biology. 6 (9): R75. doi:10.1186/gb-2005-6-9-r75.
- ↑ Wadler, C. S.; Vanderpool, C. K. (27 November 2007). "A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide". Proceedings of the National Academy of Sciences. 104 (51): 20454–20459. doi:10.1073/pnas.0708102104. PMC 2154452.
- ↑ Johnsson, Per; Lipovich, Leonard; Grandér, Dan; Morris, Kevin V. (March 2014). "Evolutionary conservation of long non-coding RNAs; sequence, structure, function". Biochimica et Biophysica Acta (BBA) - General Subjects. 1840 (3): 1063–1071. doi:10.1016/j.bbagen.2013.10.035. PMC 3909678.
- ↑ Freyhult, E. K.; Bollback, J. P.; Gardner, P. P. (6 December 2006). "Exploring genomic dark matter: A critical assessment of the performance of homology search methods on noncoding RNA". Genome Research. 17 (1): 117–125. doi:10.1101/gr.5890907. PMC 1716261.
- ↑ Nawrocki, E. P.; Eddy, S. R. (4 September 2013). "Infernal 1.1: 100-fold faster RNA homology searches". Bioinformatics. 29 (22): 2933–2935. doi:10.1093/bioinformatics/btt509. PMC 3810854.
- ↑ Eddy, SR; Durbin, R (11 June 1994). "RNA sequence analysis using covariance models". Nucleic Acids Research. 22 (11): 2079–88. doi:10.1093/nar/22.11.2079. PMC 308124. PMID 8029015.
- 1 2 "Weblogo". UC Berkeley. Retrieved 30 December 2017.
- ↑ "Clustal FAQ #Symbols". Clustal. Retrieved 8 December 2014.
- ↑ "ECR Browser". ECR Browser. Retrieved 9 January 2018.
- ↑ Earl, Dent; Nguyen, Ngan; Hickey, Glenn; Harris, Robert S.; Fitzgerald, Stephen; Beal, Kathryn; Seledtsov, Igor; Molodtsov, Vladimir; Raney, Brian J.; Clawson, Hiram; Kim, Jaebum; Kemena, Carsten; Chang, Jia-Ming; Erb, Ionas; Poliakov, Alexander; Hou, Minmei; Herrero, Javier; Kent, William James; Solovyev, Victor; Darling, Aaron E.; Ma, Jian; Notredame, Cedric; Brudno, Michael; Dubchak, Inna; Haussler, David; Paten, Benedict (December 2014). "Alignathon: a competitive assessment of whole-genome alignment methods". Genome Research. 24 (12): 2077–2089. doi:10.1101/gr.174920.114.
- ↑ Rouli, L.; Merhej, V.; Fournier, P.-E.; Raoult, D. (September 2015). "The bacterial pangenome as a new tool for analysing pathogenic bacteria". New Microbes and New Infections. 7: 72–85. doi:10.1016/j.nmni.2015.06.005.
- ↑ Méric, Guillaume; Yahara, Koji; Mageiros, Leonardos; Pascoe, Ben; Maiden, Martin C. J.; Jolley, Keith A.; Sheppard, Samuel K.; Bereswill, Stefan (27 March 2014). "A Reference Pan-Genome Approach to Comparative Bacterial Genomics: Identification of Novel Epidemiological Markers in Pathogenic Campylobacter". PLoS ONE. 9 (3): e92798. doi:10.1371/journal.pone.0092798.
- ↑ Cooper, G. M. (17 June 2005). "Distribution and intensity of constraint in mammalian genomic sequence". Genome Research. 15 (7): 901–913. doi:10.1101/gr.3577405.
- ↑ http://mendel.stanford.edu/SidowLab/downloads/gerp/. Missing or empty
|title=(help) - ↑ Pollard, K. S.; Hubisz, M. J.; Rosenbloom, K. R.; Siepel, A. (26 October 2009). "Detection of nonneutral substitution rates on mammalian phylogenies". Genome Research. 20 (1): 110–121. doi:10.1101/gr.097857.109.
- ↑ http://compgen.cshl.edu/phast/index.php. Missing or empty
|title=(help) - ↑ Fan, Xiaodan; Zhu, Jun; Schadt, Eric E; Liu, Jun S (2007). "Statistical power of phylo-HMM for evolutionarily conserved element detection". BMC Bioinformatics. 8 (1): 374. doi:10.1186/1471-2105-8-374.
- ↑ Bejerano, G. (28 May 2004). "Ultraconserved Elements in the Human Genome". Science. 304 (5675): 1321–1325. doi:10.1126/science.1098119.
- ↑ Siepel, A. (1 August 2005). "Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes". Genome Research. 15 (8): 1034–1050. doi:10.1101/gr.3715005.
- ↑ Harmston, N.; Baresic, A.; Lenhard, B. (11 November 2013). "The mystery of extreme non-coding conservation". Philosophical Transactions of the Royal Society B: Biological Sciences. 368 (1632): 20130021–20130021. doi:10.1098/rstb.2013.0021.
- ↑ Faircloth, B. C.; McCormack, J. E.; Crawford, N. G.; Harvey, M. G.; Brumfield, R. T.; Glenn, T. C. (9 January 2012). "Ultraconserved Elements Anchor Thousands of Genetic Markers Spanning Multiple Evolutionary Timescales". Systematic Biology. 61 (5): 717–726. doi:10.1093/sysbio/sys004.
- ↑ Faircloth, Brant C.; Branstetter, Michael G.; White, Noor D.; Brady, Seán G. (May 2015). "Target enrichment of ultraconserved elements from arthropods provides a genomic perspective on relationships among Hymenoptera". Molecular Ecology Resources. 15 (3): 489–501. doi:10.1111/1755-0998.12328.
- ↑ Reneker, J.; Lyons, E.; Conant, G. C.; Pires, J. C.; Freeling, M.; Shyu, C.-R.; Korkin, D. (10 April 2012). "Long identical multispecies elements in plant and animal genomes". Proceedings of the National Academy of Sciences. 109 (19): E1183–E1191. doi:10.1073/pnas.1121356109.
- ↑ Isenbarger, Thomas A.; Carr, Christopher E.; Johnson, Sarah Stewart; Finney, Michael; Church, George M.; Gilbert, Walter; Zuber, Maria T.; Ruvkun, Gary (14 October 2008). "The Most Conserved Genome Segments for Life Detection on Earth and Other Planets". Origins of Life and Evolution of Biospheres. 38 (6): 517–533. doi:10.1007/s11084-008-9148-z.
- ↑ Harris, J. K. (12 February 2003). "The Genetic Core of the Universal Ancestor". Genome Research. 13 (3): 407–412. doi:10.1101/gr.652803.
- ↑ Ban, Nenad; Beckmann, Roland; Cate, Jamie HD; Dinman, Jonathan D; Dragon, François; Ellis, Steven R; Lafontaine, Denis LJ; Lindahl, Lasse; Liljas, Anders; Lipton, Jeffrey M; McAlear, Michael A; Moore, Peter B; Noller, Harry F; Ortega, Joaquin; Panse, Vikram Govind; Ramakrishnan, V; Spahn, Christian MT; Steitz, Thomas A; Tchorzewski, Marek; Tollervey, David; Warren, Alan J; Williamson, James R; Wilson, Daniel; Yonath, Ada; Yusupov, Marat (February 2014). "A new system for naming ribosomal proteins". Current Opinion in Structural Biology. 24: 165–169. doi:10.1016/j.sbi.2014.01.002.
- ↑ Gadagkar, Sudhindra R.; Rosenberg, Michael S.; Kumar, Sudhir (15 January 2005). "Inferring species phylogenies from multiple genes: Concatenated sequence tree versus consensus gene tree". Journal of Experimental Zoology Part B: Molecular and Developmental Evolution. 304B (1): 64–74. doi:10.1002/jez.b.21026.
- ↑ Ludwig, W; Schleifer, KH (October 1994). "Bacterial phylogeny based on 16S and 23S rRNA sequence analysis". FEMS Microbiology Reviews. 15 (2–3): 155–73. doi:10.1111/j.1574-6976.1994.tb00132.x. PMID 7524576.
- ↑ Hug, Laura A.; Baker, Brett J.; Anantharaman, Karthik; Brown, Christopher T.; Probst, Alexander J.; Castelle, Cindy J.; Butterfield, Cristina N.; Hernsdorf, Alex W.; Amano, Yuki; Ise, Kotaro; Suzuki, Yohey; Dudek, Natasha; Relman, David A.; Finstad, Kari M.; Amundson, Ronald; Thomas, Brian C.; Banfield, Jillian F. (11 April 2016). "A new view of the tree of life". Nature Microbiology. 1 (5): 16048. doi:10.1038/nmicrobiol.2016.48.
- ↑ Zhang, Liqing; Li, Wen-Hsiung (February 2004). "Mammalian Housekeeping Genes Evolve More Slowly than Tissue-Specific Genes". Molecular Biology and Evolution. 21 (2): 236–239. doi:10.1093/molbev/msh010.
- ↑ Clermont, O.; Bonacorsi, S.; Bingen, E. (1 October 2000). "Rapid and Simple Determination of the Escherichia coli Phylogenetic Group". Applied and Environmental Microbiology. 66 (10): 4555–4558. doi:10.1128/AEM.66.10.4555-4558.2000.
- ↑ Kullberg, Morgan; Nilsson, Maria A.; Arnason, Ulfur; Harley, Eric H.; Janke, Axel (August 2006). "Housekeeping Genes for Phylogenetic Analysis of Eutherian Relationships". Molecular Biology and Evolution. 23 (8): 1493–1503. doi:10.1093/molbev/msl027.
- ↑ Schoch, C. L.; Seifert, K. A.; Huhndorf, S.; Robert, V.; Spouge, J. L.; Levesque, C. A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P. W.; Miller, A. N.; Wingfield, M. J.; Aime, M. C.; An, K.-D.; Bai, F.-Y.; Barreto, R. W.; Begerow, D.; Bergeron, M.-J.; Blackwell, M.; Boekhout, T.; Bogale, M.; Boonyuen, N.; Burgaz, A. R.; Buyck, B.; Cai, L.; Cai, Q.; Cardinali, G.; Chaverri, P.; Coppins, B. J.; Crespo, A.; Cubas, P.; Cummings, C.; Damm, U.; de Beer, Z. W.; de Hoog, G. S.; Del-Prado, R.; Dentinger, B.; Dieguez-Uribeondo, J.; Divakar, P. K.; Douglas, B.; Duenas, M.; Duong, T. A.; Eberhardt, U.; Edwards, J. E.; Elshahed, M. S.; Fliegerova, K.; Furtado, M.; Garcia, M. A.; Ge, Z.-W.; Griffith, G. W.; Griffiths, K.; Groenewald, J. Z.; Groenewald, M.; Grube, M.; Gryzenhout, M.; Guo, L.-D.; Hagen, F.; Hambleton, S.; Hamelin, R. C.; Hansen, K.; Harrold, P.; Heller, G.; Herrera, C.; Hirayama, K.; Hirooka, Y.; Ho, H.-M.; Hoffmann, K.; Hofstetter, V.; Hognabba, F.; Hollingsworth, P. M.; Hong, S.-B.; Hosaka, K.; Houbraken, J.; Hughes, K.; Huhtinen, S.; Hyde, K. D.; James, T.; Johnson, E. M.; Johnson, J. E.; Johnston, P. R.; Jones, E. B. G.; Kelly, L. J.; Kirk, P. M.; Knapp, D. G.; Koljalg, U.; Kovacs, G. M.; Kurtzman, C. P.; Landvik, S.; Leavitt, S. D.; Liggenstoffer, A. S.; Liimatainen, K.; Lombard, L.; Luangsa-ard, J. J.; Lumbsch, H. T.; Maganti, H.; Maharachchikumbura, S. S. N.; Martin, M. P.; May, T. W.; McTaggart, A. R.; Methven, A. S.; Meyer, W.; Moncalvo, J.-M.; Mongkolsamrit, S.; Nagy, L. G.; Nilsson, R. H.; Niskanen, T.; Nyilasi, I.; Okada, G.; Okane, I.; Olariaga, I.; Otte, J.; Papp, T.; Park, D.; Petkovits, T.; Pino-Bodas, R.; Quaedvlieg, W.; Raja, H. A.; Redecker, D.; Rintoul, T. L.; Ruibal, C.; Sarmiento-Ramirez, J. M.; Schmitt, I.; Schussler, A.; Shearer, C.; Sotome, K.; Stefani, F. O. P.; Stenroos, S.; Stielow, B.; Stockinger, H.; Suetrong, S.; Suh, S.-O.; Sung, G.-H.; Suzuki, M.; Tanaka, K.; Tedersoo, L.; Telleria, M. T.; Tretter, E.; Untereiner, W. A.; Urbina, H.; Vagvolgyi, C.; Vialle, A.; Vu, T. D.; Walther, G.; Wang, Q.-M.; Wang, Y.; Weir, B. S.; Weiss, M.; White, M. M.; Xu, J.; Yahr, R.; Yang, Z. L.; Yurkov, A.; Zamora, J.-C.; Zhang, N.; Zhuang, W.-Y.; Schindel, D. (27 March 2012). "Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi". Proceedings of the National Academy of Sciences. 109 (16): 6241–6246. doi:10.1073/pnas.1117018109.
- ↑ Man, S. M.; Kaakoush, N. O.; Octavia, S.; Mitchell, H. (26 March 2010). "The Internal Transcribed Spacer Region, a New Tool for Use in Species Differentiation and Delineation of Systematic Relationships within the Campylobacter Genus". Applied and Environmental Microbiology. 76 (10): 3071–3081. doi:10.1128/AEM.02551-09.
- ↑ Ranjard, L.; Poly, F.; Lata, J.-C.; Mougel, C.; Thioulouse, J.; Nazaret, S. (1 October 2001). "Characterization of Bacterial and Fungal Soil Communities by Automated Ribosomal Intergenic Spacer Analysis Fingerprints: Biological and Methodological Variability". Applied and Environmental Microbiology. 67 (10): 4479–4487. doi:10.1128/AEM.67.10.4479-4487.2001.
- ↑ Bidet, Philippe; Barbut, Frédéric; Lalande, Valérie; Burghoffer, Béatrice; Petit, Jean-Claude (June 1999). "Development of a new PCR-ribotyping method for based on ribosomal RNA gene sequencing". FEMS Microbiology Letters. 175 (2): 261–266. doi:10.1111/j.1574-6968.1999.tb13629.x.
- ↑ Ala, Ugo; Piro, Rosario Michael; Grassi, Elena; Damasco, Christian; Silengo, Lorenzo; Oti, Martin; Provero, Paolo; Di Cunto, Ferdinando; Tucker-Kellogg, Greg (28 March 2008). "Prediction of Human Disease Genes by Human-Mouse Conserved Coexpression Analysis". PLoS Computational Biology. 4 (3): e1000043. doi:10.1371/journal.pcbi.1000043.
- ↑ Pandey, U. B.; Nichols, C. D. (17 March 2011). "Human Disease Models in Drosophila melanogaster and the Role of the Fly in Therapeutic Drug Discovery". Pharmacological Reviews. 63 (2): 411–436. doi:10.1124/pr.110.003293.
- ↑ Huang, Hui; Winter, Eitan E; Wang, Huajun; Weinstock, Keith G; Xing, Heming; Goodstadt, Leo; Stenson, Peter D; Cooper, David N; Smith, Douglas; Albà, M Mar; Ponting, Chris P; Fechtel, Kim (2004). Genome Biology. 5 (7): R47. doi:10.1186/gb-2004-5-7-r47. Missing or empty
|title=(help) - ↑ Ge, Dongliang; Fellay, Jacques; Thompson, Alexander J.; Simon, Jason S.; Shianna, Kevin V.; Urban, Thomas J.; Heinzen, Erin L.; Qiu, Ping; Bertelsen, Arthur H.; Muir, Andrew J.; Sulkowski, Mark; McHutchison, John G.; Goldstein, David B. (16 August 2009). "Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance". Nature. 461 (7262): 399–401. doi:10.1038/nature08309.
- ↑ Bertram, L. (2009). "Genome-wide association studies in Alzheimer's disease". Human Molecular Genetics. 18: R137–R145. doi:10.1093/hmg/ddp406.
- ↑ Kellis, Manolis; Patterson, Nick; Endrizzi, Matthew; Birren, Bruce; Lander, Eric S. (15 May 2003). "Sequencing and comparison of yeast species to identify genes and regulatory elements". Nature. 423 (6937): 241–254. doi:10.1038/nature01644.
- ↑ Marchler-Bauer, A.; Lu, S.; Anderson, J. B.; Chitsaz, F.; Derbyshire, M. K.; DeWeese-Scott, C.; Fong, J. H.; Geer, L. Y.; Geer, R. C.; Gonzales, N. R.; Gwadz, M.; Hurwitz, D. I.; Jackson, J. D.; Ke, Z.; Lanczycki, C. J.; Lu, F.; Marchler, G. H.; Mullokandov, M.; Omelchenko, M. V.; Robertson, C. L.; Song, J. S.; Thanki, N.; Yamashita, R. A.; Zhang, D.; Zhang, N.; Zheng, C.; Bryant, S. H. (24 November 2010). "CDD: a Conserved Domain Database for the functional annotation of proteins". Nucleic Acids Research. 39 (Database): D225–D229. doi:10.1093/nar/gkq1189.