C-H-bindingsactivering
Koolstof-waterstofactivering, vaak ook C-H activering genoemd, kan op de simpelste manier beschreven worden als het verbreken van een binding tussen koolstof en waterstof.[1]
Vaak wordt het gebruik de term beperkt tot reacties waarbij door een organometaalverbindingen een complex gevormd wordt met een koolwaterstof. De complexvorming, met een koolstof-metaalbinding, kan beperkt blijven tot de overgangstoestand of een isoleerbaar intermediair omvatten.[2][3] Belangrijk bij deze benadering van het begrip is dat gedurende de splitsing van de C−H-binding de alkylrest aan het metaal gecoördineerd blijft.
Theoretische studies, en - belangrijker - praktisch uitgevoerde reacties, tonen aan dat de C−H-binding, traditioneel als niet-reactief beschouwd, selectief verbroken kan worden onder invloed van complexvorming. Het ontwerpen en ontwikkelen van nieuwe reagentia en katalysatoren voor de activering van de koolstof-waterstofbinding heeft de laatste jaren veel aandacht gekregen. Een belangrijke drijfveer voor dit onderzoek is de hoop in ruime hoeveelheden beschikbare en goedkope alkanen te kunnen omzetten in duurdere, lastig te maken, gefunctionaliseerde organische verbindingen.
Historisch overzicht
- 1902
De eerste, hoewel niet iedereen het daarover eens is, activeringsreactie van de C−H-binding wordt toegeschreven aan Otto Dimroth, die in 1902 de reactie beschreef van benzeen met kwik(II)acetaat (zie: organokwikchemie).[4] - 1965
De eerste C−H-activeringsreactie waarover geen meningsverschil bestaat werd door Joseph Chatt in 1965 beschreven:[5] de inschuiving van een rutheniumatoom, gecomplexeerd door dmpe, in een van de C−H-bindingen van naftaleen. In 1969 beschreef A.E. Shilov de reactie tussen zwaar water en methaan waarbij isotoop-scrambling optrad tussen de reactanten. De reactie vond plaats onder invloed van kaliumtetrachloroplatinaat. In het voorgestelde reactiemechanisme bindt methaan aan Pt(II). In een vergelijkbare reactie werd in 1972 door de groep rond Shilov methanol en chloormethaan gevonden in de reactie van een stoichiometrische hoeveelheid kaliumtetrachloroplatinaat, een katalytische hoeveelheid kaliumhexachloroplatinaat, methaan en water. Ten gevolge van het feit dat Shivov in de USSR werkte en publiceerde in de periode van de koude oorlog werd zijn werk in het westen nauwelijks opgemerkt. Tegenwoordig is dit zogenaamde Shilov-systeem een van de weinige katalysatoren voor de omzetting van alkanen in gefunctionaliseerde verbindingen.[2] - 1970
Niet alleen het puur bij elkaar voegen van reagentia leverde resultaten. Ook onder invloed van licht kan C−H-activering optreden. M. L. H. Green beschreef in 1970 de fotochemische reactie van wolfraam (als Cp2WH2 complex) met een C−H-binding van benzeen[6] en in 1979 kon George M. Whitesides als eerste een intramoleculaire reactie beschrijven van een alifatische C−H activering[7] - 1982
De volgende doorbraak werd, onafhankelijk van elkaar, door twee groepen gerapporteerd: de fotochemische activering van de C−H-binding van een volledig alifatisch systeem: Janowicz en Bergman werken met cyclohexaan en neopentaan, met de vorming van een alkylhydridometaalcomplex als resultaat: Cp*Ir(PMe3)H(C6H5). Cp* is de in de organometaalchemie gebruikelijke notatie voor de pentamethylcyclopentadienylligand.[8]
Dezelfde koolwaterstoffen werden door Graham gebruikt met het complex Cp*Ir(CO)2, waarbij iridiumhydridocomplexen ontstonden.[9]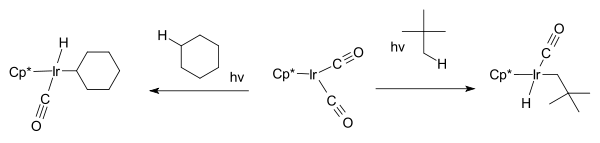
Het eerste deel van de reactie is een oxidatieve koppeling: het waterstofatoom wordt aan het alkaan onttrokken en aan het metaal gekoppeld. - 2000
Hartwig beschreef in 2000 de sterk regioselectieve borering van alkanen en arenen onder invloed van een rodiumcomplex. Bij gebruik van alkanen werd het booratoom slechts op de eindstandige plaats geïntroduceerd.[10]
:
Toepassingen
In 2007 wordt de synthese van 1-joodpentaan beschreven.[11] De standaardsynthese van dit type verbindingen verloopt via een vrije radicaalreactie, wat voor de uitkomst van de verschillende isomeren wil zeggen dat die weinig te sturen is, en bovendien dan een voorkeur voor de niet-eindstandige posities laat zien. Het alkaan pentaan wordt door het wolfraamcomplex selectief omgezet in 1-joodpentaan.
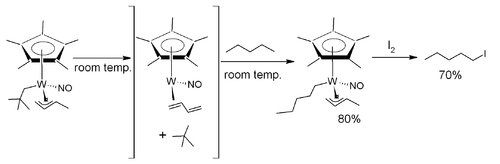
In het wolfraamcomplex treden Cp*, nitrosyl, een 3η-methylallyl en een neopentyl-ligand op. Het complex is tamelijk instabiel, en al tijdens het oplossen in pentaan bij kamertemperatuur gaat de neopentylligand verloren - deze neemt een proton over van pentaan, dat vervolgens als ligand aan wolfraam wordt gebonden. De protonuitwiseling verloopt via een 16-elkektron-intermediair, waarbij butadieen, afkomstg van het methylallyl-systeem, een belangrijke rol speelt. In een afzonderlijke reactiestap wordt bij -60°Cjood toegevoegd, waarna 1-joodpentaan ontstaat.
Activering van aromatische C−H-bindingen door metaalcomplexen is, hoewel deze koolstof-waterstofbinding meestal nog minder reactief is dan zijn alifatische tegenhanger, ook beschreven. Een voorbeeld is beschreven in de Murai-olefinekoppeling.[12] Een ander voorbeeld is de C−H-activering in N,N-dimethylbenzylamine in een rutheniumgekatalyseerde cyclometalleringsreactie[13]:

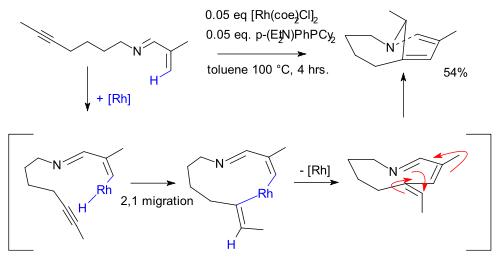
De activering van een C−H-binding in een alkeen wordt gedemonstreerd met een rodiumkatalysator in de synthese van 8,10-dimethyl-1-azabicyclo[4,3,1]deca-6,8-dieen. Vanuit structuurtheoretisch oogpunt is dit enamine interessant door het optreden van een dubbele binding aan het bruggehoofd-koolstofatoom en de relatief kleine (9 atomen) ring met de trans-binding.[14]
Reactieomstandigheden
Veel reacties waarbij activering van de C−H-binding een rol speelt, worden uitgevoerd onder tamelijk extreme omstandigheden: hoge temperatuur, sterk zuur, of sterke base, sterke oxidatoren, etc. waardoor de reacties technisch een stuk minder aantrekkelijk worden. De laatste voorbeelden laten echter ook zien dat langzaamaan ook reacties bekend worden waarbij de omstandigheden een stuk minder grofstoffelijk zijn. Hierdoor wordt de technische, en vooral economische, realisatie van de reacties een stuk makkelijker.[15]
Zie ook
Bronnen, noten en/of referenties
|