Multiple system atrophy
| Multiple system atrophy | |
|---|---|
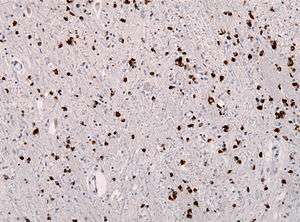 | |
| Alpha synuclein immunohistochemistry showing many glial inclusions | |
| Specialty |
Neurology |
Multiple system atrophy (MSA), also known as Shy–Drager syndrome, is a rare neurodegenerative disorder[1] characterized by tremors, slow movement, muscle rigidity, and postural instability (collectively known as parkinsonism) due to dysfunction of the autonomic nervous system, and ataxia. This is caused by progressive degeneration of neurons in several parts of the brain including the substantia nigra, striatum, inferior olivary nucleus, and cerebellum.
Many people affected by multiple system atrophy experience dysfunction of the autonomic nervous system, which commonly manifests as orthostatic hypotension, impotence, loss of sweating, dry mouth and urinary retention and incontinence. Palsy of the vocal cords is an important and sometimes initial clinical manifestation of the disorder.
The cause of MSA is uncertain and no specific risk factor has been identified,[2] although research indicates that a prion form of the alpha-synuclein protein may cause the disease.[3] About 55% of MSA cases occur in men, with typical age of onset in the late 50s to early 60s.[4] MSA often presents with some of the same symptoms as Parkinson's disease. However, those with MSA generally show little response to the dopamine medications used to treat Parkinson's disease, and only about 9% of MSA patients with tremor had a true parkinsonian pill-rolling tremor.[5]
MSA is distinct from multisystem proteinopathy, a more common muscle wasting syndrome. It should also not be confused with multiple organ dysfunction syndrome (sometimes referred to as multiple organ failure), or with multiple organ system failure (an often-fatal complication of septic shock or other very severe illnesses or injuries).
Classification
MSA, Parkinson's disease, the Lewy body dementias, and other more rare conditions make up the synucleinopathies—neurodegenerative diseases that are characterized by an abnormal accumulation of alpha-synuclein protein in the brain.[6]
Many terms have historically been used to refer to this disorder, based on the predominant systems presented. These include olivopontocerebellar atrophy (OPCA), Shy–Drager syndrome (SDS), and striatonigral degeneration (SND), which were once considered to be separate disorders.[7]
These terms and their distinctions have been dropped in recent (1996 onwards) medical usage[8] and replaced with MSA and its subtypes, but are helpful to understanding the older literature about this disease:
| Historical Name | Characteristics | Modern name and abbreviation |
| Striatonigral degeneration | predominating Parkinson's-like symptoms | MSA-P, "p" = parkinsonian subtype |
| Sporadic olivopontocerebellar atrophy (OPCA) | characterized by progressive ataxia (an inability to coordinate voluntary muscular movements) of the gait and arms and dysarthria (difficulty in articulating words) | MSA-C, "c" = cerebellar dysfunction subtype |
| Shy–Drager syndrome | characterized by Parkinsonism plus a more pronounced failure of the autonomic nervous system.[9] | No modern equivalent – this terminology fell out of favour[10] and was not specified in the 2007 consensus paper.[11] The earlier consensus of 1998[12] referred to MSA-A, "a" = autonomic dysfunction subtype but this subtype is no longer used. |
The current terminology and diagnostic criteria for the disease were established at a 2007 conference of experts on the disease and set forth in the "Second consensus statement on the diagnosis of multiple system atrophy."[13]
The Second Consensus Statement defines two categories of MSA, based on the predominant symptoms of the disease at the time of evaluation. These are:
- MSA with predominant parkinsonism (MSA-P). MSA-P is defined as MSA where extrapyramidal features predominate. The term striatonigral degeneration, parkinsonian variant, is sometimes used for this category of MSA.
- MSA with cerebellar features (MSA-C). MSA-C is defined as MSA where cerebellar ataxia predominates. It is sometimes termed sporadic olivopontocerebellar atrophy.
Signs and symptoms
MSA is characterized by the following, which can be present in any combination:[14][15]
- autonomic dysfunction
- parkinsonism (muscle rigidity +/ tremor and slow movement)
- ataxia (Poor coordination / unsteady walking)
A variant with combined features of MSA and Lewy body dementia may also exist.[16] There have also been occasional instances of frontotemporal lobar degeneration associated with MSA.[17]
Initial presentation
The most common first sign of MSA is the appearance of an "akinetic-rigid syndrome" (i.e. slowness of initiation of movement resembling Parkinson's disease) found in 62% at first presentation. Other common signs at onset include problems with balance (cerebellar ataxia) found in 22% at first presentation, followed by genito-urinary problems (9%). For men, the first sign can be erectile dysfunction (inability to achieve or sustain an erection). Women have also reported reduced genital sensitivity.[18] Both men and women often experience problems with their bladders including urgency, frequency, incomplete bladder emptying, or an inability to pass urine (retention). About 1 in 5 MSA patients will fall in their first year of disease.[4]
Progression
As the disease progresses one of three groups of symptoms predominate. These are:
- Parkinsonism (slow, stiff movement, writing becomes small and spidery)
- Cerebellar dysfunction (difficulty coordinating movement and balance)
- Autonomic nervous system dysfunction (impaired automatic body functions) including:
- postural or orthostatic hypotension, resulting in dizziness or fainting upon standing up
- urinary incontinence or urinary retention
- impotence
- constipation
- vocal cord paralysis
- dry mouth and skin
- trouble regulating body temperature due to sweating deficiency in all parts of the body
- loud snoring, abnormal breathing or inspiratory stridor during sleep
- other sleep disorders including sleep apnea, REM behavior disorder[19]
Other symptoms such as double vision can occur.[20] Not all patients experience all of these symptoms.
Some patients (20% in one study) experience significant cognitive impairment as a result of MSA.[21]
Genetics
One study found a correlation between the deletion of genes in a specific genetic region and the development of MSA in a group of Japanese patients.[22] The region in question includes the SHC2 gene which, in mice and rats, appears to have some function in the nervous system. The authors of this study hypothesized that there may be a link between the deletion of the SHC2 and the development of MSA. (See Copy-number variation for a general discussion of gene copy deletion and the variation in the number of copies of one or more sections of the DNA.)
A follow-up study was unable to replicate this finding in American MSA patients.[23] The authors of the U.S. study concluded that "Our results indicate that SHC2 gene deletions underlie few, if any, cases of well-characterized MSA in the US population. This is in contrast to the Japanese experience reported by Sasaki et al., likely reflecting heterogeneity of the disease in different genetic backgrounds."
Pathophysiology
Multiple system atrophy can be explained as cell loss and gliosis or a proliferation of astrocytes in damaged areas of the central nervous system. This damage forms a scar which is then termed a glial scar.[24] The presence of these inclusions (also known as Papp–Lantos bodies) in the movement, balance, and autonomic-control centres of the brain are the defining histopathologic hallmark of MSA. Recent studies have shown that the major filamentous component of glial and neuronal cytoplasmic inclusions is alpha-synuclein.[25] Mutations in this substance may play a role in the disease.[26] Tau proteins have been found in some GCIs.[27]
A study in 2015 suggests a new type of prion from the protein called alpha-synuclein, may be a causal agent for the disease.[28][29][30]
Diagnosis
Diagnosis of MSA can be challenging because there is no test that can definitively make or confirm the diagnosis in a living patient. Clinical diagnostic criteria were defined in 1998[12] and updated in 2007.[11] Certain signs and symptoms of MSA also occur with other disorders, such as Parkinson's disease, making the diagnosis more difficult.[31][32][33]
Both MRI and CT scanning frequently show a decrease in the size of the cerebellum and pons in those with cerebellar features. The putamen is hypodense on T2-weighted MRI and may show an increased deposition of iron in Parkinsonian form. In cerebellar form, a "hot cross" sign has been emphasized; it reflects atrophy of the pontocerebellar fibers that manifest in T2 signal intensity in atrophic pons.
A definitive diagnosis can only be made pathologically on finding abundant glial cytoplasmic inclusions in the central nervous system.[34]
Management
There is no known cure for MSA and management is primarily supportive.
Ongoing care from a neurologist specializing in "movement disorders" is recommended as the complex symptoms of MSA are often not familiar to less-specialized health care professionals.
One particularly serious problem, the drop in blood pressure upon standing up (with risk of fainting and thus injury from falling) often responds to fludrocortisone, a synthetic mineralocorticoid.[35] Another common drug treatment is midodrine (an alpha-agonist).[35] Non-drug treatments include "head-up tilt" (elevating the head of the whole bed by about 10 degrees), salt tablets or increasing salt in the diet, generous intake of fluids, and pressure (elastic) stockings. Avoidance of triggers of low blood pressure (such as hot weather, alcohol, and dehydration) are crucial.[36]
Hospice/homecare services can be very useful as disability progresses.
Levodopa (L-Dopa), a drug used in the treatment of Parkinson's disease, improves parkinsonian symptoms in a small percentage of MSA patients. A recent trial reported that only 1.5% of MSA patients experienced a less than 50% improvement when taking levodopa, and even this was a transient effect lasting less than one year. Poor response to L-Dopa has been suggested as a possible element in the differential diagnosis of MSA from Parkinson's disease.
A November, 2008 study conducted in Europe failed to find an effect for the drug riluzole in treating MSA or PSP.[4]
Rehabilitation
Management by rehabilitation professionals (physiatrists, physiotherapists, occupational therapists, speech therapists, and others) for problems with walking/movement, daily tasks, and speech problems is essential.
Physiotherapy can help to maintain the patient’s mobility and will help to prevent contractures.[24] Instructing patients in gait training will help to improve their mobility and decrease their risk of falls.[37] A physiotherapist may also prescribe mobility aids such as a cane or a walker to increase the patient’s safety.[37] Other ways a physiotherapist can help to improve the patient’s safety are to teach them to move and transfer from sitting to standing slowly to decrease risk of falls and limit the effect of postural hypotension.[37] Instruction in ankle pumping helps to return blood in the legs to the systemic circulation.[37] To further control the postural hypotension, raising the head of the bed by 8 in (20.3 cm) while sleeping may be indicated as well as the use of elastic compression garments.[14]
Speech and language therapists may assist in assessing, treating and supporting speech (dysarthria) and swallowing difficulties (dysphagia). Early intervention of swallowing difficulties is particularly useful to allow for discussion around tube feeding further in the disease progression. At some point in the progression of the disease, fluid and food modification may be suggested. Speech changes mean that alternative communication may be needed, for example communication aids or word charts.
Social workers and occupational therapists can also help with coping with disability through the provision of equipment and home adaptations, services for caregivers and access to healthcare services, both for the person with MSA as well as family caregivers.
Prognosis
MSA usually progresses more quickly than Parkinson's disease.[38] There is no remission from the disease. The average remaining lifespan after the onset of symptoms in patients with MSA is 7.9 years.[38] Almost 80% of patients are disabled within five years of onset of the motor symptoms, and only 20% survive past 12 years.[39] Rate of progression differs in every case and speed of decline may vary widely in individual patients.
O’Sullivan and colleagues (2008) identified early autonomic dysfunction to be the most important early clinical prognostic feature regarding survival in MSA. Patients with concomitant motor and autonomic dysfunction within three years of symptom onset had a shorter survival duration, in addition to becoming wheelchair dependent and bed-ridden at an earlier stage than those who developed these symptoms after three years from symptom onset. Their study also showed that when patients with early autonomic dysfunction develop frequent falling, or wheelchair dependence, or severe dysphagia, or require residential care, there is a shorter interval from this point to death.[40]
Epidemiology
The rate of MSA is estimated at 4.6 cases per 100,000 people.[38][41] This disease is more common in men than in women, with studies showing ratios ranging from between 1.4:1[24] to ratios as high as 1.9:1.[14] Chef Kerry Simon died from complications of MSA.
Research
A July, 2012, study suggested that mesenchymal stem cell therapy could delay the progression of neurological deficits in patients with MSA-cerebellar type, suggesting the potential of mesenchymal stem cell therapy as a treatment candidate of MSA.[42]
References
- ↑ "multiple system atrophy" at Dorland's Medical Dictionary
- ↑ "National Study Seeks Cause of Baffling, Fatal Disorder Called Multiple System Atrophy". UCSD Health Sciences Communications Healthbeat. December 5, 2003. Retrieved 2008-07-01.
- ↑ Prusiner SB, Giles K, et al. (2015-07-22). "Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism". PNAS. 112: E5308–17. doi:10.1073/pnas.1514475112. PMC 4586853. PMID 26324905.
- 1 2 3 Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN (2008). "Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: The NNIPPS Study". Brain. 132 (Pt 1): 156–71. doi:10.1093/brain/awn291. PMC 2638696. PMID 19029129.
- ↑ "Multiple System Atrophy Clinical Presentation". Retrieved January 7, 2018.
- ↑ Goedert M, Jakes R, Spillantini MG (2017). "The Synucleinopathies: Twenty Years On". J Parkinsons Dis (Review). 7 (s1): S53–S71. doi:10.3233/JPD-179005. PMC 5345650. PMID 28282814.
- ↑ Ahmed, Z.; Asi, YT.; Sailer, A.; Lees, AJ.; Houlden, H.; Revesz, T.; Holton, JL. (Feb 2012). "The neuropathology, pathophysiology and genetics of multiple system atrophy". Neuropathol Appl Neurobiol. 38 (1): 4–24. doi:10.1111/j.1365-2990.2011.01234.x. PMID 22074330.
- ↑ The Consensus Committee of the American Autonomic Society and the American Academy of Neurology (1996). "Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy". Neurology. 46 (5): 1470. doi:10.1212/wnl.46.5.1470. PMID 8628505.
- ↑ Shy GM, Drager GA (1960). "A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study". Arch. Neurol. 2: 511–27. doi:10.1001/archneur.1960.03840110025004. PMID 14446364.
- ↑ Schatz, IJ (Jul 1, 1996). "Farewell to the "Shy–Drager syndrome"". Annals of Internal Medicine. 125 (1): 74–5. doi:10.7326/0003-4819-125-1-199607010-00012. PMID 8644992.
- 1 2 Gilman, S; Wenning, GK; Low, PA; Brooks, DJ; Mathias, CJ; Trojanowski, JQ; Wood, NW; Colosimo, C; Dürr, A; Fowler, CJ; Kaufmann, H; Klockgether, T; Lees, A; Poewe, W; Quinn, N; Revesz, T; Robertson, D; Sandroni, P; Seppi, K; Vidailhet, M (Aug 26, 2008). "Second consensus statement on the diagnosis of multiple system atrophy". Neurology. 71 (9): 670–6. doi:10.1212/01.wnl.0000324625.00404.15. PMC 2676993. PMID 18725592.
- 1 2 Gilman, S; Low, PA; Quinn, N; Albanese, A; Ben-Shlomo, Y; Fowler, CJ; Kaufmann, H; Klockgether, T; Lang, AE; Lantos, PL; Litvan, I; Mathias, CJ; Oliver, E; Robertson, D; Schatz, I; Wenning, GK (Feb 1, 1999). "Consensus statement on the diagnosis of multiple system atrophy". Journal of the Neurological Sciences. 163 (1): 94–8. doi:10.1016/s0022-510x(98)00304-9. PMID 10223419.
- ↑ Gillman, S.; Wenning, GK; Low, PA; Brooks, DJ; Mathias, CJ; Trojanowski, JQ; Wood, NW; Colosimo, C; et al. (2008). "Second consensus statement on the diagnosis of multiple system atrophy". Neurology. 71 (9): 670–6. doi:10.1212/01.wnl.0000324625.00404.15. PMC 2676993. PMID 18725592.
- 1 2 3 Swan L, Dupont J (May 1999). "Multiple system atrophy". Phys Ther. 79 (5): 488–94. PMID 10331752.
- ↑ Burn DJ, Jaros E (December 2001). "Multiple system atrophy: cellular and molecular pathology". Mol. Pathol. 54 (6): 419–26. PMC 1187133. PMID 11724918.
- ↑ Sikorska B, Papierz W, Preusser M, Liberski PP, Budka H (2017). "Synucleinopathy with features of both multiple system atrophy and dementia with Lewy bodies". Neuropathology and Applied Neurobiology. 33 (1): 126–9. doi:10.1111/j.1365-2990.2006.00817.x. PMID 17239015.
- ↑ Aoki N1, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, Weiner M, Lipton A, Powers JM, White CL 3rd, Dickson DW. (July 2015). "Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with α-synuclein". Acta Neuropathol. 130 (1): 93–105. doi:10.1007/s00401-015-1442-z.
- ↑ Oertel WH1, Wächter T, Quinn NP, Ulm G, Brandstädter D. (Apr 2003). "Reduced genital sensitivity in female patients with multiple system atrophy of parkinsonian type". Mov Disord. 18 (4): 430–2. doi:10.1002/mds.10384. PMID 12671951.
- ↑ Gilman S, Koeppe RA, Chervin RD, et al. (July 2003). "REM sleep behavior disorder is related to striatal monoaminergic deficit in MSA". Neurology. 61 (1): 29–34. doi:10.1212/01.wnl.0000073745.68744.94. PMID 12847152.
- ↑ NINDS NIH MSA with Orthostatic Hypotension
- ↑ Brown RG, Lacomblez L, Landwehrmeyer BG, Bak T, Uttner I, Dubois B, Agid Y, Ludolph A, Bensimon G, Payan C, Leigh NP; for the NNIPPS Study Group (August 2010). "Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy". Brain. 133 (Pt 8): 2382–93. doi:10.1093/brain/awq158. PMID 20576697.
- ↑ Sasaki H, Emi M, Iijima H, et al. (2011). "Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy". Mol Brain. 4: 24. doi:10.1186/1756-6606-4-24. PMC 3141657. PMID 21658278.
- ↑ Ferguson, MC; Garland, EM; Hedges, L; Womack-Nunley, B; Hamid, R; Phillips JA, 3rd; Shibao, CA; Raj, SR; Biaggioni, I; Robertson, D (Feb 2014). "SHC2 gene copy number in multiple system atrophy (MSA)". Clinical Autonomic Research. 24 (1): 25–30. doi:10.1007/s10286-013-0216-8. PMC 3946192. PMID 24170347.
- 1 2 3 Wenning GK, Colosimo C, Geser F, Poewe W (February 2004). "Multiple system atrophy". Lancet Neurol. 3 (2): 93–103. doi:10.1016/S1474-4422(03)00662-8. PMID 14747001.
Wenning GK, Colosimo C, Geser F, Poewe W (March 2004). "Erratum". Lancet Neurol. 3 (3): 137. doi:10.1016/S1474-4422(04)00695-7. - ↑ Arima K, Uéda K, Sunohara N, Arakawa K, Hirai S, Nakamura M, Tonozuka-Uehara H, Kawai M (November 1998). "NACP/alpha-synuclein immunoreactivity in fibrillary components of neuronal and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy". Acta Neuropathol. 96 (5): 439–44. doi:10.1007/s004010050917. PMID 9829806.
- ↑ Al-Chalabi, A; Dürr, A; Wood, NW; Parkinson, MH; Camuzat, A; Hulot, JS; Morrison, KE; Al-Chalabi A, Dürr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS, Morrison KE, Renton A, Sussmuth SD, Landwehrmeyer BG, Ludolph A, Agid Y, Brice A, Leigh PN, Bensimon G; NNIPPS Genetic Study Group.; et al. (Sep 2009). Heutink, Prof. Peter, ed. "Genetic Variants of the α-Synuclein Gene SNCA Are Associated with Multiple System Atrophy". PLoS ONE. 4 (9): e7114. doi:10.1371/journal.pone.0007114. PMC 2743996. PMID 19771175.
- ↑ Piao YS, Hayashi S, Hasegawa M, Wakabayashi K, Yamada M, Yoshimoto M, Ishikawa A, Iwatsubo T, Takahashi H (Mar 2001). "Co-localization of alpha-synuclein and phosphorylated tau in neuronal and glial cytoplasmic inclusions in a patient with multiple system atrophy of long duration". Acta Neuropathol. 101 (3): 285–93. doi:10.1007/s004010000292. PMID 11307630.
- ↑ New Type of Prion May Cause, Transmit Neurodegeneration. Nicholas Weiler. August 31, 2015.
- ↑ Another Fatal Brain Disease May Come from the Spread of 'Prion' Proteins. Wired Science, Rachael Rettner. August 31, 2015.
- ↑ Prusinera, Stanley B.; Woerman, Amanda L.; Gilesa, Kurt; et al. (July 22, 2015). "Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism". PNAS USA. 112: E5308–17. doi:10.1073/pnas.1514475112. PMC 4586853. PMID 26324905. Retrieved 2015-09-05.
- ↑ Vanderbilt Autonomic Dysfunction Cente. "Multiple System Atrophy / Shy Drager Syndrome". Retrieved May 29, 2010.
- ↑ "multiple system atrophy overview".
- ↑ Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, Wszolek ZK, Langston JW, Dickson DW. (August 4, 2015). "When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients". Neurology. 85 (5): 404–12. doi:10.1212/WNL.0000000000001807. PMC 4534078. PMID 26138942.
- ↑ Papp MI, Kahn JE, Lantos PL (Dec 1989). "Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy–Drager syndrome)". J Neurol Sci. 94 (1–3): 79–100. doi:10.1016/0022-510X(89)90219-0. PMID 2559165.
- 1 2 Multiple system atrophy (MSA) mayoclinic.org, accessed 20 May 2018
- ↑ Aminoff MJ, Greenberg DA, Simon RP. "Chapter 7. Movement Disorders". Clinical Neurology (6th ed.).
- 1 2 3 4 Hardy, Joanne (2008). "Multiple system atrophy: pathophysiology, treatment and nursing care". Nursing Standard. 22 (22): 50–56. doi:10.7748/ns2008.02.22.22.50.c6359. PMID 18333558.
- 1 2 3 Bower J, Maraganore D, McDonnell S, Rocca W (1997). "Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990". Neurology. 49 (5): 1284–8. doi:10.1212/wnl.49.5.1284. PMID 9371909.
- ↑ Watanabe H, Saito Y, Terao S, et al. (May 2002). "Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients". Brain. 125 (Pt 5): 1070–83. doi:10.1093/brain/awf117. PMID 11960896.
- ↑ O`Sullivan, S. S.; Massey, L. A.; Williams, D. R.; Silveira-Moriyama, L.; Kempster, P. A.; Holton, J. L.; Revesz, T.; Lees, A. J. (2008). "Clinical outcomes of progressive supranuclear palsy and multiple system atrophy". Brain. 131 (5): 1362–72. doi:10.1093/brain/awn065. PMID 18385183.
- ↑ Prevalence of rare diseases : Bibliographic data (PDF) (Report). Orphanet. November 2008. p. 20. Retrieved 2009-01-19.
- ↑ Lee, Phil Hyu; Lee, Ji E.; Kim, Han-Soo; Song, Sook K.; Lee, Hye Sun; Nam, Hyo Suk; Cheong, June-Won; Jeong, Yong; Park, Hae-Jeong; Kim, Dong Joon; Nam, Chung Mo; Lee, Jong Doo; Kim, Hyun Ok; Sohn, Young H. (2012). "A randomized trial of mesenchymal stem cells in multiple system atrophy". Annals of Neurology. 72 (1): 32–40. doi:10.1002/ana.23612. PMID 22829267.
External links
- Medical Textbook: "Multiple System Atrophy" edited by Gregor Wenning and Alessandra Fanciulli
- The Multiple System Atrophy Coalition (formerly The SDS/MSA Support Group, Inc.), a US based non-profit for people with Multiple System Atrophy
- The Multiple System Atrophy Trust founded by Sarah Matheson, a UK registered charity providing information about MSA.
- Movement Disorder Society MSA Study Group, an administrative framework for global collaborative MSA research
- Autonomic Dysfunction Center at Vanderbilt University
- The European MSA Study Group, an Innsbruck-based European MSA Study Group comprising 25 academic centres of excellence dedicated to MSA research
- Diary of a MSA-patient.
| Classification | |
|---|---|
| External resources |