Epidermolysis bullosa
Epidermolysis bullosa[1] (afgekort EB) is een erfelijke, vooralsnog ongeneeslijke huidziekte waarbij blaren op de huid ontstaan. De aandoening is gekenmerkt door defecten in de eiwitten die de opperhuid (epidermis) aan de lederhuid (dermis) bevestigen. De epidermis sluit cellulair niet goed aan op de dermis — zogenaamde celbrugjes ontbreken — waardoor hij bij lichte wrijving al snel loskomt van de lederhuid met vorming van blaren waarin zich sereus vocht bevindt. Bij gezonde mensen ontstaan deze blaren alleen na langdurige wrijving op de huid, zoals voetblaren bij lange wandelingen. Maar bij mensen met ernstige EB ontstaan blaren alleen al door het dragen van kleding en bij alledaagse aanrakingen, zoals knuffelen of het optillen van een baby. In zelfhulpliteratuur wordt ook het woord “vlinderhuid” (van een “vlinderkind”) gebruikt.[2]
Epidermolysis bullosa
| ||||
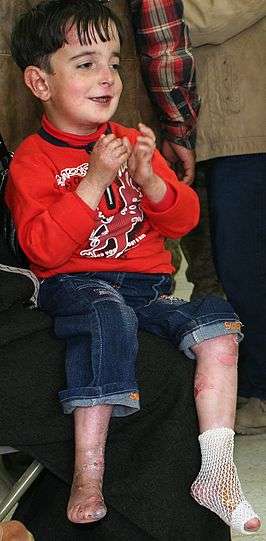 | ||||
Een vijfjarige jongen met congenitale epidermolysis bullosa | ||||
| Coderingen | ||||
| ICD-10 | Q81 | |||
| ICD-9 | 757.39 | |||
| eMedicine | derm/124 | |||
| MeSH | D004820 | |||
| ||||
| Neem het voorbehoud bij medische informatie in acht. Raadpleeg bij gezondheidsklachten een arts. |
Indeling
Bij EB kan men drie verschijningsvormen onderscheiden, die elk weer varianten vertonen, die onderwerp van onderzoek blijven. De drie verschijningsvormen van EB zijn:
- Epidermolysis bullosa simplex[1] (70%): blaarvorming ontstaat in de basale cellaag. Hij wordt veroorzaakt door (dominante) mutaties in keratine 5 of keratine 14.
- Epidermolysis bullosa dystrophicans[1] (25%): blaarvorming onder de lamina densa van de basale membraan. Hij wordt veroorzaakt door mutaties in het gen collageen VII. Er bestaat een milde (dominante) en ernstige (recessieve) variant.
- Epidermolysis bullosa junctionalis (5%): blaarvorming ter plaatse van de lamina lucida van de basale membraan. Hij wordt veroorzaakt door meestal recessieve mutaties in de genen voor laminine 5 of collageen XVII.
Dit is een percentsgewijze indeling volgens de wereldwijd vastgestelde frequentie.
Bij de medisch-klinische indeling wisselen de epidermolysis bullosa dystrophicans en epidermolysis bullosa junctionalis van plaats: die is gebaseerd op de uitwendig waarneembare graad van beschadiging van de huidlagen.
Het beloop van de ziekte is moeilijk voorspelbaar, maar per verschijningsvorm wel enigszins, mede afhankelijk van de verzorging die men zich eigen maakt en/of krijgt. Een EB-patiënt wordt geboren met een van de drie verschijningsvormen en die gaan, gelet op de beschrijving van het genetisch defect, niet in elkaar over. De junctionalis- en de dystroficans-recessieve EB-verschijningsvormen zijn levensbedreigend, omdat de huid het grootste menselijke orgaan is en wonden, misvormingen, littekenvorming en infectiegevaar permanent zijn.
Sociaal
- Simplex
Zoals met veel andere ziekten ervaart ook de EB-patiënt een zekere mate van uitsluiting. Kinderen worden vaak gepest, omdat de ziekte wordt onderschat: vooral de simplex-verschijningsvorm geeft daartoe aanleiding omdat blaren in de schoenen onzichtbaar zijn. Leeftijdgenoten zien de patiënt alleen manken en verwijten hem lijntrekkerij.
- Dystrophicans en junctionalis
Hoe zichtbaarder, des te meer weerstand bij de gezonde mens: men denke aan scènes bij zwembaden en verkleedhokjes. Bij confrontatie met een EB-patiënt moet met klem worden herhaald dat deze ziekte erfelijk (ofwel genetisch) is en helemaal niet besmettelijk.
Verzorging
Goede verzorging kan van een epidermolysis bullosa dystrophicans geen epidermolysis bullosa simplex maken. Aangezien EB ongeneeslijk is, is de best mogelijke verzorging het hoogst haalbare. Blaren en blaasjes van EB zijn vergelijkbaar met brandwonden van de tweede graad en moeten als zodanig worden behandeld, maar nog zorgvuldiger dan bij verbrande patiënten met een overigens gezonde huid. Immers, bij de vormen die niet simplex[3] zijn, kunnen weer nieuwe blaren ontstaan door het verschuiven van het verband en het te haastig verwijderen van het verband; pleisters zijn af te raden. Bij alle vormen vullen de blaren zich makkelijk opnieuw na doorprikken, zodat het doorprikken en de nabehandeling enkele dagen met volharding moeten worden herhaald. Vooral bij warm weer is het voor een EB-patiënt niet verrassend wanneer hij in de herstellende blaar de vorming van een nieuwe blaar vaststelt. Geregeld is er ook bloed vermengd in het sereus blaarvocht.
Terwijl de wondverzorging voor junctionalis en dystroficans patiënten permanent en noodzakelijk is (zie verder), is ze voor simplex-patiënten een jaarlijks terugkomende leerschool: herfst en winter als doorgaans blaarvrije seizoenen doen ze hun ziekte en de bijbehorende verzorging vergeten; iedereen wil zich graag gezond voelen en “erbij horen” (zoals anderen in de groep handelen zonder uitval). Maar grofweg tussen april en september ontstaan de blaren op voeten en handen bij warm weer. Daarnaast komen sporadisch blaartjes voor veroorzaakt door elastiekbanden in sokken en onderbroeken en tussen de billen bij fietstochten. Niet uit te voeren door de andere patiënten en voor simplex-patiënten af te raden activiteiten zijn: wandelingen van meer dan een 5-tal km, alle sporten die men met sportschoenen uitoefent, hanteren van scharen bij het tuinonderhoud (herhaalde drukbeweging), roeien. Met het voorbehoud dat er ook binnen EB-simplex verschillende graden bestaan, is de simplexvorm beter beheersbaar naarmate de patiënt ouder wordt en minder in beweging is. Toch heeft hij dan meer eeltvorming en de normale noodzakelijke eeltlaag remt de vorming van blaren niet af zoals bij een gezonde huid — blaren ontstaan ook onder het eelt (voornamelijk op de hielen) en duwen dit eelt omhoog. Sedert ca. 1950 zijn al veel manieren van blaarverzorging uitgeprobeerd en weer opgegeven, maar het volledig verwijderen van de blaarhuid wordt tegenwoordig in alle omstandigheden sterk afgeraden. Volgens de huidige medische praktijk heb je de keuze tussen droog makende en vochtig houdende wondheling: voor beide manieren bestaan er steriel houdende verbanden. Het grotendeels behouden van de blaarhuid maakt een veel hogere graad van steriliteit mogelijk, die vooral de eerste twee dagen na de punctie van groot belang is. De recente methode om de blaar gesloten te houden en te overplakken met een pleister die een blaarvocht absorberende gel zou bevatten, levert bij EB-blaren niet het beschreven gunstige resultaat.[4]
Bij de vormen van epidermolysis bullosa dystrophicans kunnen naast huidblaren ook elders blaren en wondjes ontstaan. Het betreft hier vooral het slijmvlies in ogen, mond, keel en slokdarm. Er kunnen blaren of wondjes aan het gebit, in de nieren en de urinewegen ontstaan. Vingers en tenen krijgen vervormingen en de nagelbedden worden ingesloten. De defecte huid is bij de geboorte direct vast te stellen.
Veruit de ernstigste vorm is de Herlitzvariant van epidermolysis bullosa junctionalis. De ouders zijn dan drager, vaak zonder dit te beseffen (recessief). Junctionalis EB is nog altijd niet tijdens de zwangerschap voorspelbaar. Deze vorm heeft een slechte prognose: de kinderen overlijden meestal binnen de eerste twee levensjaren aan de gevolgen van vernauwing van de luchtpijp: bemoeilijkte ademhaling, ondervoeding en uitputting. Karakteristiek voor dit ziektebeeld zijn de huidbeschadigingen die meteen na de geboorte meestal rond de nagels zichtbaar zijn. Op andere delen van de huid is deze huidschade pas na enige maanden vast te stellen.
Toekomst
Naast verzorging wordt er voortdurend gezocht in ziekenhuizen met het specialisme genetica naar voorkoming van EB bij de conceptie — selecteren van EB-vrije sperma- en eicellen (IVF toepassen) — en naar remedies tegen de hier beschreven defecten van de huideiwitten. EB is een van de vele kwalen die mogelijk voor stamceltherapie in aanmerking komen. Onderzoekers rekenen op nog minstens 10 jaar voor een praktische remedie wordt gevonden. Oktober 2015 werd de eerste succesvolle oplossing via gentherapie tot stand gebracht. Met behulp van een virus werd een goede kopie van het LAMB3-gen in het DNA van de huidcellen gebracht.[5]
Belangenbehartiging
Omdat een optimale verzorging het hoogst haalbare is, zijn goede raad en ontmoetingen met lotgenoten nooit weg. Dankzij de komst van het internet is ook voor EB de belangenbehartiging in een stroomversnelling geraakt. Er was in het Verenigd Koninkrijk een patiëntenvereniging-zelfhulpgroep ontstaan op initiatief van de moeder van Debra, een meisje dat in 1978 aan de ziekte overleden is. Debra is nu de naam van de internationale zelfhulpgroep die in vele landen een vertegenwoordiging heeft; enkele jaren heeft men internationaal het ervoor bedachte acroniem Dystrophic Epidermolysis Bullosa Research Association gebruikt, maar aangezien niet-dystroficans EB'ers terecht bezwaar maakten, heeft men die omschrijving nu verlaten.[6][7]
EB is als ziekte, maar niet als handicap in de ziekteverzekering opgenomen: het wordt geklasseerd als zeldzame ziekte, een vertaling van de internationale term rare disease.[8][9] Een terugbetalingsregeling door de ziekenfondsen (België) of ziekteverzekeringen (Nederland) dekt lang niet alle kosten, maar vele andere zeldzame ziekten ontberen ook een deugdelijke regeling.
Voetnoten
Externe links
|