Metal phosphine complex
In coordination chemistry phosphines are L-type ligands. Unlike most metal ammine complexes, metal phosphine complexes tend to be lipophilic, displaying good solubility in organic solvents. They also are compatible with metals in multiple oxidation states. Because of these two features, metal phosphine complexes are useful in homogeneous catalysis.[1] Prominent examples of metal phosphine complexes include Wilkinson's catalyst (Rh(PPh3)3Cl), Grubbs' catalyst, and tetrakis(triphenylphosphine)palladium(0).
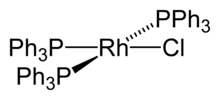
Preparation
The first phosphine complexes were cis- and trans-PtCl2(PEt3)2 reported by Cahours and Gal in 1870.[2]
A popular phosphine ligand is triphenylphosphine, a shelf-stable solid that undergoes oxidation in air relatively slowly.
Many complexes continue to be prepared from metal halides. For example, treatment of palladium chloride with triphenylphosphine yields monomeric bis(triphenylphosphine)palladium(II) chloride units.[3]
- [PdCl2]n + 2n PPh3 → n PdCl2(PPh3)2
M-PR3 bonding
Phosphines primarily function as Lewis bases, interacting with metals as σ donor ligands.
| L | ν(CO) cm−1 |
|---|---|
| P(t-Bu)3 | 2056.1 |
| PMe3 | 2064.1 |
| PPh3 | 2068.9 |
| P(OEt)3 | 2076.3 |
| PCl3 | 2097.0 |
| PF3 | 2110.8 |
Phosphine ligands are also π-acceptors. Their π-acidity arises from overlap of P-C σ* anti-bonding orbitals with filled metal orbitals. Aryl- and fluorophosphines are stronger π-acceptors than alkylphosphines. Trifluorophosphine (PF3) is a strong π-acid with bonding properties akin to those of the carbonyl ligand.[5] In early work, phosphine ligands were thought to utilize 3d orbitals to form M-P pi-bonding, but it is now accepted that d-orbitals on phosphorus are not involved in bonding.[6] The energy of the σ* orbitals is lower for phosphines with electronegative substituents, and for this reason phosphorus trifluoride is a particularly good π-acceptor.[7]
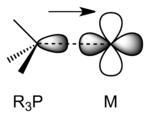 R3P–M σ bonding
R3P–M σ bonding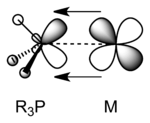 R3P–M π backbonding
R3P–M π backbonding
Steric properties
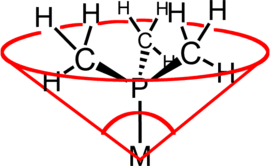
By changes in one or more of the three organic substituents, the steric and electronic properties of phosphine ligands can be manipulated. These properties are manifested in the catalytic properties of the host metal center.[8] The steric properties of phosphine ligands can be ranked by their steric properties. The Tolman cone angle is widely used.[9]
Reactivity
Phosphine ligands are usually "spectator" rather than "actor" ligands. They generally do not participate in reactions, except to dissociate from the metal center. In certain high temperature hydroformylation reactions, the scission of P-C bonds is observed however.[10] The thermal stability of phosphines ligands is enhanced when they are incorporated into pincer complexes.
Applications to homogeneous catalysis
One of the first applications of phosphine ligands in catalysis was the use of triphenylphosphine in “Reppe” chemistry (1948), which included reactions of alkynes, carbon monoxide, and alcohols.[11] In his studies, Reppe discovered that this reaction more efficiently produced acrylic esters using NiBr2(PPh3)2 as a catalyst instead of NiBr2. Shell developed cobalt-based catalysts modified with trialkylphosphine ligands for hydroformylation (now a rhodium catalyst is more commonly used for this process).[12] The success achieved by Reppe and his contemporaries led to many industrial applications.[13]
Complexes of other organophosphorus ligands
The popularity and usefulness of phosphine complexes has led to the popularization of complexes of many related organophosphorus ligands.[2] Complexes of arsines have also been widely investigated, but are avoided in practical applications because of concerns about toxicity.
Complexes of primary and secondary phosphines
Most work focuses on complexes of triorganophosphines, but primary and secondary phosphines, respectively RPH2 and R2PH, also function as ligands. Such ligands are less basic and have small cone angles. These complexes are susceptible to deprotonation leading to phosphido-bridged dimers and oligomers:
- 2 LnM(PR2H)Cl → [LnM(μ-PR2)]2 + 2 HCl
Complexes of PRx(OR')3−x
Nickel(0) complexes of phosphites, e.g., Ni[P(OEt)3]4 are useful catalysts for hydrocyanation of alkenes. Related complexes are known for phosphinites (R2P(OR')) and phosphonites (RP(OR')2).
Diphosphine complexes
Due to the chelate effect, ligands with two phosphine groups bind more tightly to metal centers than do two monodentate phosphines. The conformation of diphosphines is restricted, and for this reason many are used in asymmetric catalysis, e.g. Noyori asymmetric hydrogenation. Several diphosphines have been developed; many of these diphosphines have two arylphosphines linked by a short alkylidene bridge, e.g. 1,2-bis(diphenylphosphino)ethane (dppe). 1,1'-Bis(diphenylphosphino)ferrocene is interesting, because it uses a ferrocene unit as the linker. Several trans-spanning ligands like xantphos and spanphos are known as well. The complex dichloro(1,3-bis(diphenylphosphino)propane)nickel is useful in Kumada coupling.
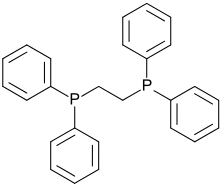 1,2-Bis(diphenylphosphino)ethane (dppe)
1,2-Bis(diphenylphosphino)ethane (dppe)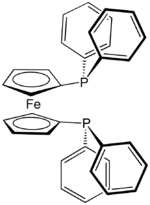 1,1'-Bis(diphenylphosphino)ferrocene (dppf)
1,1'-Bis(diphenylphosphino)ferrocene (dppf)
References
- ↑ Hartwig, J. F. Organotransition Metal Chemistry, from Bonding to Catalysis; University Science Books: New York, 2010. ISBN 1-891389-53-X
- 1 2 C. A. McAuliffe, ed. (1973). Transition Metal Complexes of Phosphorus, Arsenic, and Antimony Ligands. J. Wiley. ISBN 0-470-58117-4. .
- ↑ Norio Miyaura and Akira Suzuki (1993). "Palladium-catalyzed reaction of 1-alkenylboronates with vinylic halides: (1Z,3E)-1-Phenyl-1,3-octadiene". Organic Syntheses. ; Collective Volume, 8, p. 532
- ↑
- ↑ Orpen, A. G.; Connelly, N. G. (1990). "Structural systematics: the role of P-A σ* orbitals in metal-phosphorus π-bonding in redox-related pairs of M-PA3 complexes (A = R, Ar, OR; R = alkyl)". Organometallics. 9 (4): 1206–1210. doi:10.1021/om00118a048.
- ↑ Gilheany, D. G. (1994). "No d Orbitals but Walsh Diagrams and Maybe Banana Bonds: Chemical Bonding in Phosphines, Phosphine Oxides, and Phosphonium Ylides". Chem. Rev. 94 (5): 1339–1374. doi:10.1021/cr00029a008.
- ↑ Crabtree, Robert H. (2009). The Organometallic Chemistry of the Transition Metals (5th ed.). Wiley. pp. 99–100. ISBN 978-0-470-25762-3.
- ↑ R. H. Crabtree (2005). "4. Carbonyls, Phosphine Complexes, and Ligand Substitution Reactions". The Organometallic Chemistry of the Transition Metals (4th ed.). ISBN 0-471-66256-9.
- ↑ Tolman, C. A. (1977). "Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis". Chem. Rev. 77 (3): 313–48. doi:10.1021/cr60307a002.
- ↑ Garrou, Philip E. (1985). "Transition-metal-mediated phosphorus-carbon bond cleavage and its relevance to homogeneous catalyst deactivation". Chem. Rev. 85: 171–185. doi:10.1021/cr00067a001.
- ↑ Reppe, W.; Schweckendiek, W. J. (31 July 1948). "Cyclisierende Polymerisation von Acetylen. III Benzol, Benzolderivate und hydroaromatische Verbindungen". Justus Liebigs Annalen der Chemie. 560 (1): 104–116. doi:10.1002/jlac.19485600104.
- ↑ Slaugh, L; Mullineaux, R. (1968). "Novel Hydroformylation catalysts". J. Organomet. Chem. 13 (2): 469. doi:10.1016/S0022-328X(00)82775-8.
- ↑ P. W.N.M. van Leeuwen "Homogeneous Catalysis: Understanding the Art, 2004 Kluwer, Dordrecht. ISBN 1-4020-2000-7