Arginine:glycine amidinotransferase deficiency
| Arginine:glycine amidinotransferase deficiency | |
|---|---|
 | |
| Classification and external resources | |
| ICD-10 | E72.9 |
| OMIM | 612718 |
| DiseasesDB | 36670 |
| GeneReviews | |
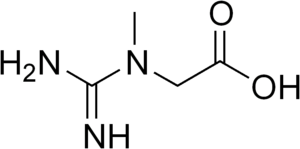
Arginine:glycine amidinotransferase deficiency (AGAT deficiency) is an autosomal recessive cerebral creatine deficiency caused by a deficiency of the enzyme arginine:glycine amidinotransferase. This enzyme deficiency results in decreased creatine synthesis, and is caused by biallelic pathogenic variants in GATM. Individuals with AGAT deficiency are intellectually disabled and have muscle weakness. The symptoms of AGAT deficiency are caused by the lack of creatine in specific tissues, most notably muscle and brain. Oral creatine supplementation can be used to treat AGAT deficiency, with early intervention providing the best results. All creatine deficiencies are rare, there have been fewer than 20 individuals reported in medical literature with AGAT deficiency. This disorder was first described in 2000.
Signs and symptoms
As with other cerebral creatine deficiency syndromes, individuals affected with AGAT deficiency are intellectually disabled and can have seizures.[1] These symptoms are caused by the lack of creatine in skeletal muscles and in the brain. AGAT deficiency was first identified in 2000, in a pair of sisters aged 4 and 6. Both sisters had severe intellectual disability.[2]
Genetics
.png)
AGAT deficiency is caused by deficient activity of arginine:glycine amidinotransferase, which is coded for by GATM, located on the long arm of chromosome 15. AGAT deficiency is inherited in an autosomal recessive manner, which means pathogenic variants must be inherited from each parent. This enzyme catalyzes the first step in creatine biosynthesis, the combination of arginine and glycine to form guanidinoacetate, which also results in the formation or ornithine as a by product. These reactions take place primarily in the kidney and pancreas.
Diagnosis
AGAT deficiency can be suspected from clinical findings, although there is significant phenotypic overlap with the most common presenting symptoms of intellectual disability and muscle weakness. Biochemical testing of plasma and urine will show decreased levels of creatine and guanidinoacetate. Magnetic resonance spectroscopy (MRS) of the brain will also show an absence of creatine, which is normally present. This finding is not specific to AGAT deficiency, it can be observed in all three cerebral creatine deficiencies. The combination of biochemical testing and MRS findings can be strongly suggestive of AGAT deficiency. Confirmation would most often be done with molecular testing of GATM. Identification of biallelic pathogenic variants in GATM would be confirmation of a diagnosis of AGAT deficiency.[1] Uncertain findings on molecular testing may be able to be confirmed by enzyme assays, or by measuring creatine uptake in fibroblasts.[1]
Treatment
The main focus of treatment for AGAT deficiency is supplementation of creatine, with the goal of replenishing cerebral creatine levels. This is done with oral creatine supplementation. Treatment is most effective if it is started early in life, before symptoms are apparent. Treatment in affected individuals does not reverse intellectual disability or improve cognitive function. For treatment at any age,even if intellectual disability was present, all individuals showed improvement in muscle weakness.[1] In an asymptomatic sibling, who was started on treatment due to the earlier diagnosis of an affected sibling, early intervention with creatine supplementation resulted in improved outcomes when compared to their untreated siblings at the same age.[1]
References
- 1 2 3 4 5 "Creatine Deficiency Syndrome". Gene Reviews. National Institutes of Health. Retrieved 2018-09-26.
- ↑ "CEREBRAL CREATINE DEFICIENCY SYNDROME 3; CCDS3". Johns Hopkins University. Retrieved 2018-10-11.