Photochlorination
Photochlorination is a chemical reaction which is initiated by light, in which either hydrogen is replaced by chlorine in a hydrocarbon compound or chlorine is reacted via an addition reaction to an aromatic or olefinic hydrocarbon. Photochlorination is carried out in addition to thermal and catalytic chlorination on an industrial scale, usually in the liquid phase, sometimes in the presence of inert solvents. The process is exothermic and proceeds as a chain reaction initiated by the homolytic cleavage of molecular chlorine into chlorine radicals by ultraviolet radiation.
The chlorinated hydrocarbons produced via photochlorination are often only industrial intermediates and react with a large number of basic chemicals to secondary products such as alcohols, mercaptans, amines and carboxylic acids. The chemical industry uses low molecular weight chlorinated compounds, such as tetrachloromethane, as solvents. Higher molecular-weight chloroalkanes serve as insecticides, as flame retardants or as plasticizers in plastics and coatings. Chlorinated hydrocarbons are also used as intermediates in the chemical industry for the production of silicones or detergents.
History

Chlorination is one of the oldest known substitution reactions in chemistry. The French chemist Jean-Baptiste Dumas already investigated the substitution of hydrogen for chlorine by acetic acid in candle wax as early as 1830.[1] He showed that for each mole of chlorine introduced into a hydrocarbon, one mole of hydrogen chloride is formed and noted the light-sensitivity of this reaction.[2]
The first work on the influence of light on the rate of chemical reactions is given by Theodor Grotthuss. In 1819 Grotthuss published a paper on the chemical effectiveness of light and formulated the photochemical absorption law. Following it, in a physico-chemical system only the absorbed fraction of the incident radiation produces an effect in this system; reflected and transmitted radiation has no effect.[3]
The work of Max Planck, published in 1900, showed that light consists of discrete quanta.[4] The excitation of a single chemical reaction by a light quantum could be explained, but not the quantum yield of reactions such as photochlorination. The idea that these reactions might be chain reactions was brought up by Max Bodenstein (1913). He assumed that in the reaction of two molecules not only the end product of the reaction can be formed, but also unstable, reactive intermediates which can continue the chain reaction.[5]
Chemists studied the reaction in detail because of the importance of the reaction to the understanding of chemistry, of substitution patterns and derivatives. However, photochlorination could only be transferred to chemical industry when, at the end of the nineteenth century, cheap chlorine was available from chloralkali electrolysis.[6]
Chlorinated alkanes found an initial application in pharyngeal sprays. These contained chlorinated alkanes in relatively large quantities as solvents for chloramine T from 1914 to 1918. The Sharpless Solvents Corporation commissioned the first industrial photochloration plant for the chlorination of pentane in 1929.[7] The commercial production of chlorinated paraffins for use as high-pressure additives in lubricants began around 1930.[8] Around 1935 the process was technically stable and commercially successful.[7] However, it was only in the years after World War II that a greater build-up of photochloration capacity began. In 1950, the United States produced more than 800,000 tons of chlorinated paraffin hydrocarbons. The major products were ethyl chloride, tetrachlorocarbon and dichloromethane.[9] Because of concerns about health and environmentally relevant problems such as the ozone depletion behavior of light volatile chlorine compounds, the chemical industry developed alternative procedures that did not require chlorinated compounds. As a result of the following replacement of chlorinated by non-chlorinated products, worldwide production volumes have declined considerably over the years.[8][10]
Reactions
Substitution reactions
The substitution of the hydrogen atoms in a hydrocarbon is fully random, with tertiary hydrogen atoms reacting faster than secondary ones reacting faster than primary ones. At 30 °C the relative reaction rates of primary, secondary and tertiary hydrogen atoms are in a relative ratio of approximately 1 to 3.25 to 4.43.[11]
Upon radiation the reaction involves alkyl and chlorine radicals following a chain reaction according to the given scheme:





Chain termination occurs by recombination of chlorine radicals to molecular chlorine on the vessel wall.[12] Impurities such as oxygen (present in electrochemically obtained chlorine) also cause chain termination.
In photochlorination there is no rearrangement of the carbon chain. All possible mono- and multi-chlorinated compounds are formed.[13] However, the monosubstituted hydrocarbons are the main products of photochlorination. The formation of multi-chlorinated products can be reduced to a certain degree by working with a high excess of hydrocarbons or by diluting the chlorine with nitrogen.
The selectivity of photochlorination (with regard to substitution of primary, secondary or tertiary hydrogens) can be controlled by the interaction of the chlorine radical with the solvent, such as benzene, tert-butylbenzene or carbon disulfide.[14] The complex formation between benzene and the chloride radical reduces its reactivity which increases the selectivity.[15] By varying the solvent the ratio of primary to secondary hydrogens can be tailored to ratios between 1: 3 to 1: 31.[16] At higher temperatures, the reaction rates of primary, secondary and tertiary hydrogen atoms equallize. Therefore, photochlorination is usually carried out at lower temperatures.[13]



Kinetics

Reactants of the photochlorination can be both gaseous and liquid hydrocarbons. In the case of liquid starting materials, chlorine is introduced upon stirring. Reactions in the gas phase, such as the photochlorination of methane, are also possible in principle. However, the reaction enthalpy must in this case mostly be absorbed by the specific heat capacities of the gases involved, what limits the conversion.[18]
In general, it is necessary to bring the reactants close to the light source in order to obtain the highest possible luminous efficacy. For this purpose, the reaction mixture can be irradiated either directly or in a flow-through side arm of a reactor with a suitable light source. Gaseous hydrocarbons are introduced with chlorine into an inert solvent and reacted there under irradiation.[19]
A disadvantage of photochemical processes is the low efficiency of the conversion of electrical energy in the radiation energy of the required wavelength. In addition to the radiation, light sources generate plenty of heat, which in turn requires cooling energy. In addition, most light sources emit polychromatic light, even though only monochromatic light is needed.[20] A high quantum yield, however, compensates for these disadvantages. The quantum yield for the photochlorination of n-heptane is about 7000, for example.[21] In photochlorination plants, the quantum yield is about 100. In contrast to the thermal chlorination, which can utilize the formed reaction energy, the energy required to maintain the photochemical reaction must be constantly delivered.[19]
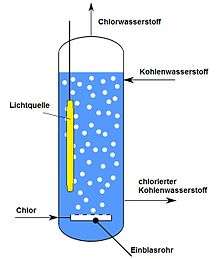
The presence of inhibitors, such as oxygen or nitrogen oxides, must be avoided. Too high chlorine concentrations lead to high absorption near the light source and have a disadvantageous effect.[16] Working at low temperatures is advantageous since side reactions are avoided (as the selectivity is increased) and the yield is increased (since gaseous reactants are driven out less from the solvent).
The starting materials can sometimes be cooled before the reaction to such an extent that the reaction heat is absorbed without further cooling of the mixture. In the case of gaseous or low-boiling starting materials, work under overpressure is necessary. Due to the large number of possible raw materials, a large number of processes have been described.[22][23] Photochlorination is usually carried out in a stirred tank reactor, a bubble column reactor or a tube reactor, followed by further processing depending on the target product.[24] In case of a stirred tank reactor, the lamp (generally shaped as an elongated cylinder) is provided with a cooling jacket and placed in the reaction solution. Tube reactors are made from quartz or glass tubes, which are irradiated from the outside. Using a stirred tank reactor has the advantage that no light is lost to the environment. However, the intensity of light drops rapidly with the distance to the light source due to adsorption by the reactants.[19]
The influence of the radiation on the reaction rate can often be represented by a power law based on the quantum flow density, i.e. the mole light quantum (previously measured in the unit Einstein) per area and time. One objective in the design of reactors is therefore to determine the economically most favorable dimensioning with regard to an optimization of the quantum current density.[25]
Products
Chlorinated products can be converted into other intermediates and end products by a variety of reactions, for example by hydrolysis in alcohols or by reaction with alkali metal cyanides in nitriles (which can be hydrolyzed with water to form carboxylic acids or reduced to hydrogen with amines). By conversion with metallic magnesium in Grignard reactions, carbon skeletons can be synthesized from the intermediate alkyl-magnesium halides.[26] In Friedel-Crafts alkylations, chloroalkanes are used to prepare alkylaromatics.[27]
Chlorinated paraffins
By photochlorination chloroparaffins can be prepared from alkanes. Compared to thermal chlorination, the risk of the formation of secondary products by thermolysis (for example, by elimination of hydrogen chloride) is very small. The selectivity is low because the reaction takes place by a radical mechanism. Mixtures of complex composition consisting of several chlorinated paraffins are formed. The degree of chlorination varies and the exact composition of the resulting product mixtures is often not known.[8] In 1985 the world production was 300,000 tonnes; since then the production volumes are falling in Europe and North America.[28] In China, on the other hand, production rose sharply. China produced more than 600,000 tonnes of chlorinated paraffins in 2007, while in 2004 it was less than 100,000 tonnes.[29]
Chlorinated paraffins have the general sum formula CxH(2x−y+2)Cly and are categorized into three groups: Low molecular weight chlorinated paraffins are short chain chloroparaffins (SCCP) with 10 to 13 carbon atoms, followed by medium chain chloroparaffins (MCCP) with carbon chain lengths of 14 to 17 carbon atoms and long chain chlorinated paraffins (LCCP), owing a carbon chainwith more than 17 carbon atoms. Approximately 70% of the chloroparaffins produced are MCCPs with a degree of chlorination from 45 to 52%. The remaining 30% are divided equally between SCCP and LCCP.[8] Short chain chloroparaffins have high toxicity and easily accumulate in the environment. The European Union has classified SCCP as a category III carcinogen and restricted its use.[30]
Benzyl chloride, benzal chloride and benzotrichloride
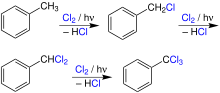
The photochlorination of the side-chain of toluene generates its mono- to trichlorinated products, the most important of which is the mono-substituted benzyl chloride. It is converted via hydrolysis into benzyl alcohol, which is used as an intermediate for the production of plasticizers. Benzyl chloride can also be converted via benzyl cyanide with subsequent hydrolysis into phenylacetic acid.[31][32] The disubstituted benzal chloride is used as a raw material for the production of benzaldehyde. As a pure substance, benzaldehyde is used to impart almond smell to foods.[33] Furthermore, benzaldehyde is used as an intermediate for the production of malachite green and other dyes.[34] The trisubstituted benzotrichloride is used for the hydrolysis of the synthesis of benzoyl chloride:[35]


By reaction with alcohols, benzoyl chloride can be converted into the corresponding esters. With sodium peroxide it turns into dibenzoyl peroxide, a radical initiator for polymerizations. However, the atom economy of these syntheses is poor, since stoichiometric amounts of salts are obtained.
Chloromethanes
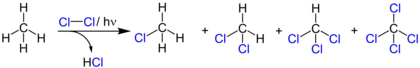
An example of photochlorination at low temperatures and under ambient pressure is the chlorination of chloromethane to dichloromethane. The liquefied chloromethane (boiling point -24 °C) is mixed with chlorine in the dark and then irradiated with a mercury-vapor lamp. The resulting dichloromethane has a boiling point of 41 °C and is later separated by distillation from methyl chloride.[36]
The photochlorination of methane has a lower quantum yield than the chlorination of dichloromethane. Due to the high light intensity required, the intermediate products are directly chlorinated, so that mainly tetrachloromethane is formed.[36]
Process variants
Sulfochlorination
The sulfochlorination first described by Cortes F. Reed in 1936 proceeds under almost identical conditions as the conventional photochlorination. [48] In addition to chlorine, sulfur dioxide is also introduced into the reaction mixture. The products formed are alkylsulfonyl chlorides, which are further processed into surfactants.[37]
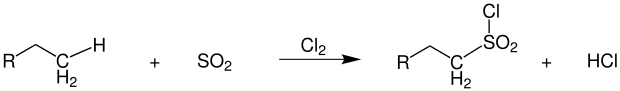
Hydrochloric acid is formed as a coupling product, as is the case with photo chlorination. Since direct sulfonation of the alkanes is hardly possible, this reaction has proven to be useful. Due to chlorine, which is bound directly to the sulfur, the resulting products are highly reactive. As secondary products there are alkyl chlorides formed by pure photochlorination, as well as several sulfochlorinated products in the reaction mixture.[38] photochlorination.
Photobromination

Photobromination with elemental bromine proceeds analogous to photochlorination also via a radical mechanism. In the presence of oxygen, the hydrogen bromide formed is partly oxidised back to bromine, resulting in an increased yield.[39] Because of the easier dosage of the elemental bromine and the higher selectivity of the reaction, photobromination is preferred over photochlorination at laboratory scale. For industrial applications, bromine is usually too expensive (as it is present in sea water in small quantities only and produced from oxidation with chlorine).[40][41] Instead of elemental bromine, N-bromosuccinimide is also suitable as a brominating agent.[42] The quantum yield of photobromination is usually much lower than that of photochlorination.
References
General sources: [43][44][45][46][47] [48][49][50][51][52] [53][54]
- Jean-Baptiste Dumas: Ueber die Einwirkung des Chlors auf den aus essigsauren Salzen entstehenden Kohlenwasserstoff. In: Annalen der Chemie und Pharmacie. 33, 1840, p. 187–189, doi:10.1002/jlac.18400330205.
- Jean-Baptiste Dumas: Über das Gesetz der Substitution und die Theorie der Typen, Lieb. Ann., Vol. 33, 1840, p. 259–300.
- Theodor Von Grotthuss: Auszug aus vier Abhandlungen Physikalisch-chemischen Inhalts. In: Annalen der Physik und der physikalischen Chemie. 61, 1819, p. 50–74, doi:10.1002/andp.18190610105.
- Max Planck: Zur Theorie des Gesetzes der Energieverteilung im Normalspectrum. In: Verhandlungen der Deutschen physikalischen Gesellschaft. 2, Nr. 17, 1900, S. 245, Berlin (vorgetragen am 14. Dezember 1900; PDF Archived 2015-08-07 at the Wayback Machine).
- Max Bodenstein: Photochemische Kinetik des Chlorknallgases. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie, 19, 1913, p. 836–856, doi:10.1002/bbpc.19130192104.
- Franz Rudolf Minz, Reinhard Schliebs: Moderne Verfahren der Großchemie: Chlor und Natronlauge. In: Chemie in unserer Zeit. 12. Jahrg. 1978, Nr. 5, ISSN 0009-2851, p. 135–145.
- Wilhelm Hirschkind: Chlorination of Saturated Hydrocarbons. In: Industrial & Engineering Chemistry. 41, 1949, p. 2749–2752, doi:10.1021/ie50480a021.
- United Nations Environment Programme, International Labour Organisation, World Health Organisation, International Programme on Chemical Safety, Environmental Health Criteria 181: CHLORINATED PARAFFINS.
- Earl T. McBee, Ogden R Pierce: Halogenation. In: Industrial & Engineering Chemistry. 46, 1954, p. 1835–1841, doi:10.1021/ie50537a031.
- Martin Dameris, Thomas Peter, Ulrich Schmidt, Reinhard Zellner: Das Ozonloch und seine Ursachen, In: Chemie in unserer Zeit, 2007, 41, p. 152–168; doi:10.1002/ciuz.200700418.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 978-3-13-201904-1, p. 91.
- Max Bodenstein: Sitzung vom 15. Dezember 1930. Berichte der deutschen chemischen Gesellschaft (A and B Series) 64.1 (1931): A1–A4.
- Keith U. Ingold, J. Lusztyk, K. D. Raner: The unusual and the unexpected in an old reaction. The photochlorination of alkanes with molecular chlorine in solution. In: Accounts of Chemical Research. 23, 1990, p. 219, doi:10.1021/ar00175a003.
- Glen A. Russell: Solvent Effects in the Reactions of Free Radicals and Atoms. III. Effects of Solvents in the Competitive Photochlorination of Hydrocarbons and Their Derivatives. In: Journal of the American Chemical Society. 80, 1958, p. 4997–5001, doi:10.1021/ja01551a057.
- D. J. Hurley, R. W. Rosenthal, R. C. Williamson: Effect of Chlorination Conditions on Preparation and Isomer Distribution of Linear Detergent Alkylate. In: Industrial & Engineering Chemistry Product Research and Development. 4, 1965, p. 22, doi:10.1021/i360013a007.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 978-3-13-201904-1, p. 95.
- Richard Wegler (Hrsg.): Chemie der Pflanzenschutz und Schädlingsbekämpfungsmittel. Band 1, Springer-Verlag, 1970, ISBN 978-3-642-46212-2, p. 129–132.
- Mario Schiavello (Hrsg.): Photoelectrochemistry, Photocatalysis and Photoreactors Fundamentals and Developments. Springer Netherlands, 2009, ISBN 978-90-481-8414-9, p. 564.
- Martin Fischer: Industrial Applications of Photochemical Syntheses. In: Angewandte Chemie International Edition in English. 17, 1978, p. 16–26, doi:10.1002/anie.197800161.
- Dieter Wöhrle, Michael W. Tausch, Wolf-Dieter Stohrer: Photochemie: Konzepte, Methoden, Experimente. Wiley & Sons, 1998, ISBN 978-3-527-29545-6, p. 271–275.
- Joachim Stauff, H. J. Schumacher: Apparatur zur Untersuchung von Lichtreaktionen der Halogene mit organischen Substanzen: Die Lichtreaktion zwischen Chlor und n‐Heptan. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 48, 1942, p. 271–278, doi:10.1002/bbpc.194200006.
- US Grant 1379367, F. Sparre & W. E. Masland, "Process of Chlorination", issued 1921-05-24, assigned to Du Pont
- US Grant 1459777, R. Leiser & F. Ziffer, "Process and Apparatus for the Chlorination of Methane", issued 1920-02-14, assigned to Ziffer Fritz and Leiser Richard
- David A. Mixon, Michael P. Bohrer, Patricia A. O’Hara: Ultrapurification of SiCl4 by photochlorination in a bubble column reactor. In: AIChE Journal. 36, 1990, p. 216–226, doi:10.1002/aic.690360207.
- H. Hartig: Einfache Dimensionierung, photochemischer Reaktoren. In: Chemie Ingenieur Technik – CIT. 42, 1970, p. 1241–1245, doi:10.1002/cite.330422002.
- Victor Grignard: Sur quelques nouvelles combinaisons organométalliques du magnèsium et leur application à des synthèses d’alcools et d’hydrocarbures. In: CR Hebd. Séances Acad. Sci., Ser. C, 130, 1900, p. 1322–1324, available at Gallica, dt. Über einige neue metallorganische Verbindungen von Magnesium und deren Anwendung auf Synthesen von Alkoholen und Kohlenwasserstoffen
- Charles Friedel, James Mason Crafts: Sur une nouvelle méthode générale de synthèse d’hydrocarbures, d’acétones, etc., Compt. Rend., 84: 1392 & 1450.
- Heinz Strack: Chlorinated paraffins. In: Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim, 1986, VCH Verlagsgesellschaft, Vol. A6, p. 323–330.
- Heidelore Fiedler (Hrsg.): Chlorinated Paraffins. In: The Handbook of Environmental Chemistry, Springer-Verlag, 2010, ISBN 978-3-642-10760-3, p. 8.
- "Richtlinie 2002/45/EG des Europäischen Parlaments und des Rates vom 25. Juni 2002 zur 20. Änderung der Richtlinie 76/769/EWG des Rates hinsichtlich der Beschränkungen des Inverkehrbringens und der Verwendung gewisser gefährlicher Stoffe und Zubereitungen (kurzkettige Chlorparaffine) ".
- Roger Adams, A. F. Thal: Benzyl Cyanide. In: Organic Syntheses. 2, 1922, p. 9, doi:10.15227/orgsyn.002.0009.
- Roger Adams, A. F. Thal: Phenylacetic Acid [α-Toluic acid]. In: Organic Syntheses. 2, 1922, p. 63, doi:10.15227/orgsyn.002.0063.
- Final Report on the Safety Assessment of Benzaldehyde. In: International Journal of Toxicology. 25, 2006, p. 11–27, doi:10.1080/10915810600716612.
- Siegfried Hauptmann: Organische Chemie, 2. Auflage, VEB Deutscher Verlag für Grundstoffindustrie, Leipzig, 1985, ISBN 3-342-00280-8, p. 757.
- Barbara Elvers (Hrsg.): Ullmann’s Encyclopedia of Industrial Chemistry: 7th Edition, Wiley-VCH, 2002, ISBN 978-3-527-30385-4, p. 139.
- Eugen Müller (Hrsg,), E. Forche, W. Hahn: Methoden der organischen Chemie Band V/3: Halogenverbindungen. Fluorverbindungen. Herstellung, Reaktivität und Umwandlung. Chlorverbindungen. Thieme Verlag, 1962, p. 571–573.
- US Grant 2174492, Cortes F. Reed, "Preparation of alkane sulphonyl chlorides", issued 1938-09-26, assigned to Charles L Horn
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 978-3-13-201904-1, p. 165–176.
- M. Le Blanc, K. Andrich: Photobromierung des Toluols. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 20.18‐19, 1914, p. 543–547, doi:10.1002/bbpc.19140201804.
- Klaus Schwetlick (2009). Organikum: organisch-chemisches Grundpraktikum (in German) (23rd ed.). Weinheim: Wiley-VCH. p. 206. ISBN 978-3-527-32292-3.
- Rudolf Bock: Gewinnung von Brom aus Meerwasser. In: Chemie Ingenieur Technik – CIT. 25, 1953, p. 245, doi:10.1002/cite.330250507.
- Hans-Friedrich Grützmacher, Jürgen Schmiegel: Dithia-diaza[n.2]metacyclophan-ene. In: Chemische Berichte. 122, 1989, p. 1929–1933, doi:10.1002/cber.19891221017.
- Hans Von Halban: Die Lichtabsorption des Chlors. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 28, 1922, p. 496–499, doi:10.1002/bbpc.19220282304.
- Holleman, Arnold Frederik; Wiberg, Egon (2001), Wiberg, Nils (ed.), Inorganic Chemistry, translated by Eagleson, Mary; Brewer, William, San Diego/Berlin: Academic Press/De Gruyter, p. 71, ISBN 0-12-352651-5
- Arthur John Allmand: Part I.—Einstein’s law of photochemical equivalence. Introductory address to Part I. In: Trans. Faraday Soc. 21, 1926, p. 438, doi:10.1039/TF9262100438.
- Marion L. Sharrah, Geo. C. Feighner: Synthesis of Dodecylbenzen – Synthetic Detergent Intermediate. In: Industrial & Engineering Chemistry. 46, 1954, p. 248–254, doi:10.1021/ie50530a020.
- "Richtlinie 2003/53/EG des Europäischen Parlaments und des Rates vom 18. Juni 2003 zur 26. Änderung der Richtlinie 76/769/EWG des Rates über Beschränkungen des Inverkehrbringens und der Verwendung gewisser gefährlicher Stoffe und Zubereitungen (Nonylphenol, Nonylphenolethoxylat und Zement)"..
- C. Decker, M. Balandier, J. Faure: Photochlorination of Poly(vinyl Chloride). I. Kinetics and Quantum Yield. In: Journal of Macromolecular Science: Part A – Chemistry. 16, 2006, p. 1463–1472, doi:10.1080/00222338108063248.
- Tsutomu Nakagawa, Sumio Yamada: Modification of polyolefin films by photochlorination. In: Journal of Applied Polymer Science. 16, 1972, p. 1997–2012, doi:10.1002/app.1972.070160813.
- Rolf C. Schulz, Rainer Wolf: Copolymerisation zwischen Vinylencarbonat und Isobutylvinyläther. In: Kolloid-Zeitschrift & Zeitschrift für Polymere. 220, 1967, p. 148–151, doi:10.1007/BF02085908.
- Ernst Bartholomé, Ernst Biekert, Heinrich Hellmann: Umwelt- und Arbeitsschutz. (Ullmanns Encyklopädie der technischen Chemie Bd. 6). Wiley-VCH, 4. Auflage, 1981, ISBN 978-3-527-20006-1, p. 206.
- R. Newe, P. Schmidt, K. Friese, B. Hösselbarth: Das Verfahren der strahlenchemischen Chlorierung von Polyvinylchlorid. In: Chemische Technik, 41(4), 1989, p. 141–144.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.), Otto Bayer, Hans Meerwein, Karl Ziegler: Methoden der organischen Chemie. V/3 Fluorine and Chlorine Compounds . Thieme Verlag, Stuttgart 1962, ISBN 978-3-13-203004-6, p. 524.
- US Grant 2046090, Cortes F. Reed, "Method of halogenating compounds and product resulting therefrom", issued 1936-06-30, assigned to Charles L Horn