Inverse gas chromatography
Inverse gas chromatography is a physical characterization analytical technique that is used in the analysis of the surfaces of solids.[1]

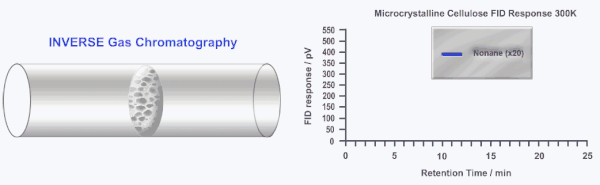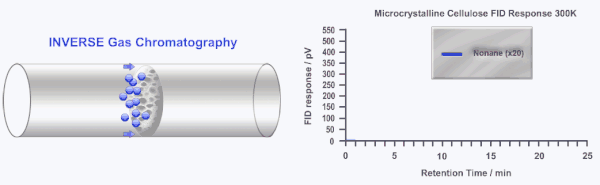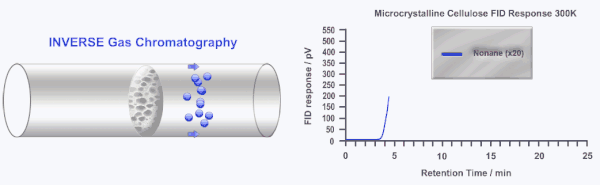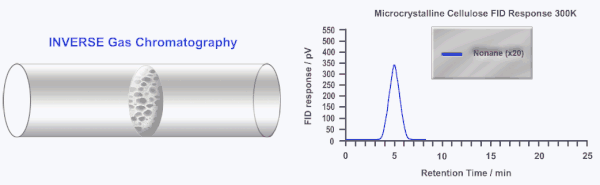
Inverse gas chromatography or IGC is a highly sensitive and versatile gas phase technique developed over 40 years ago to study the surface and bulk properties of particulate and fibrous materials. In IGC the roles of the stationary (solid) and mobile (gas or vapor) phases are inverted from traditional analytical gas chromatography (GC). In GC, a standard column is used to separate and characterize several gases and/or vapors. In IGC, a single gas or vapor (probe molecule) is injected into a column packed with the solid sample under investigation. Instead of an analytical technique, IGC is considered a materials characterization technique.
During an IGC experiment a pulse or constant concentration of a known gas or vapor (probe molecule) is injected down the column at a fixed carrier gas flow rate. The retention time of the probe molecule is then measured by traditional GC detectors (i.e. flame ionization detector or thermal conductivity detector). Measuring how the retention time changes as a function of probe molecule chemistry, probe molecule size, probe molecule concentration, column temperature, or carrier gas flow rate can elucidate a wide range of physico-chemical properties of the solid under investigation. Several in depth reviews of IGC have been published previously.[2][3]
IGC experiments are typically carried out at infinite dilution where only small amounts of probe molecule are injected. This region is also called Henry's law region or linear region of the sorption isotherm. At infinite dilution probe-probe interactions are assumed negligible and any retention is only due to probe-solid interactions. The resulting retention volume, VRo, is given by the following equation:

where j is the James–Martin pressure drop correction, m is the sample mass, F is the carrier gas flow rate at standard temperature and pressure, tR is the gross retention time for the injected probe, to is the retention time for a non-interaction probe (i.e. dead-time), and T is the absolute temperature.
Surface energy determination
The main application of IGC is to measure the surface energy of solids (fibers, particulates, and films). Surface energy is defined as the amount of energy required to create a unit area of a solid surface; analogous to surface tension of a liquid. Also, the surface energy can be defined as the excess energy at the surface of a material compared to the bulk. The surface energy (γ) is directly related to the thermodynamic work of adhesion (Wadh) between two materials as given by the following equation:

where 1 and 2 represent the two components in the composite or blend. When determining if two materials will adhere it is common to compare the work of adhesion with the work of cohesion, Wcoh = 2γ. If the work of adhesion is greater than the work of cohesion, then the two materials are thermodynamically favored to adhere.
Surface energies are commonly measured by contact angle methods. However, these methods are ideally designed for flat, uniform surfaces. For contact angle measurements on powders, they are typically compressed or adhered to a substrate which can effectively change the surface characteristics of the powder. Alternatively, the Washburn method can be used, but this has been shown to be affected by column packing, particle size, and pore geometry.[4] IGC is a gas phase technique, thus is not subject to the above limitations of the liquid phase techniques.
To measure the solid surface energy by IGC a series of injections using different probe molecules is performed at defined column conditions. It is possible to ascertain both the dispersive component of the surface energy and acid-base properties via IGC. For the dispersive surface energy, the retention volumes for a series of n-alkane vapors (i.e. decane, nonane, octane, heptanes, etc.) are measured. The Dorris and Gray.[5] or Schultz [6] methods can then be used to calculate the dispersive surface energy. Retention volumes for polar probes (i.e. toluene, ethyl acetate, acetone, ethanol, acetonitrile, chloroform, dichloromethane, etc.) can then be used to determine the acid-base characteristics of the solid using either the Gutmann,[7] or Good-van Oss theory.[8]
Other parameters accessible by IGC include: heats of sorption [1], adsorption isotherms,[9] energetic heterogeneity profiles,[10][11] diffusion coefficients,[12] glass transition temperatures [1],[13] Hildebrand [14][15] and Hansen [16] solubility parameters, and crosslink densities.[17]
Applications
IGC experiments have applications over a wide range of industries. Both surface and bulk properties obtained from IGC can yield vital information for materials ranging from pharmaceuticals to carbon nanotubes. Although surface energy experiments are most common, there are a wide range of experimental parameters that can be controlled in IGC, thus allowing the determination of a variety of sample parameters. The below sections highlight how IGC experiments are utilized in several industries.
Polymers and coatings
IGC has been used extensively for the characterization of polymer films, beads, and powders. For instance, IGC was used to study surface properties and interactions amongst components in paint formulations.[18] Also, IGC has been used to investigate the degree of crosslinking for ethylene propylene rubber using the Flory–Rehner equation [17]. Additionally, IGC is a sensitive technique for the detection and determination of first and second order phase transitions like melting and glass transition temperatures of polymers.[19] Although other techniques like differential scanning calorimetry are capable of measuring these transition temperatures, IGC has the capability of glass transition temperatures as a function of relative humidity.[20]
Pharmaceuticals
The increasing sophistication of pharmaceutical materials has necessitated the use for more sensitive, thermodynamic based techniques for materials characterization. For these reasons, IGC, has seen increased use throughout the pharmaceutical industry. Applications include polymorph characterization,[21] effect of processing steps like milling,[22] and drug-carrier interactions for dry powder formulations.[23] In other studies, IGC was used to relate surface energy and acid-base values with triboelectric charging [24] and differentiate the crystalline and amorphous phases [23].
Fibers
Surface energy values obtained by IGC have been used extensively on fibrous materials including textiles,[25] natural fibers,[26] glass fibers,[27] and carbon fibers.[28] Most of these and other related studies investigating the surface energy of fibers are focusing on the use of these fibers in composites. Ultimately, the changes in surface energy can be related to composite performance via the works of adhesion and cohesion discussed previously.
Nanomaterials
Similar to fibers, nanomaterials like carbon nanotubes, nanoclays, and nanosilicas are being used as composite reinforcement agents. Therefore, the surface energy and surface treatment of these materials has been actively studied by IGC. For instance, IGC has been used to study the surface activity of nanosilica, nanohematite, and nanogeoethite.[29] Further, IGC was used to characterize the surface of as received and modified carbon nanotubes.[30]
Metakaolins
IGC was used to characterize the adsorption surface properties of calcined kaolin (metakaolin) and the grinding effect on this material.[31]
See also
- Surface energy
- Adhesion
- Wetting
- Wetting transition
- Material characterization
- Sessile drop technique
References
- Mohammadi-Jam, S.; Waters, K.E. (2014). "Inverse gas chromatography applications: A review". Advances in Colloid and Interface Science. 212: 21–44. doi:10.1016/j.cis.2014.07.002. ISSN 0001-8686.
- J. Condor and C. Young, Physicochemical measurement by gas chromatography, John Wiley and Sons, Chichester, UK (1979)
- F. Thielmann, Journal of Chromatography A. 1037 (2004) 115.
- J. Dove, G. Buckton, and C. Doherty, International Journal of Pharmaceutics. 138 (1996) 199–206.
- G.M. Doris and D.G. Gray, Journal of Colloids and Interfacial Science. 56 (1964) 353.
- J. Schultz, L. Lavielle, and C. Martin, Journal of Adhesion. 77 (1980) 353–362.
- V. Gutmann, Coordination Chemistry Reviews. 2 (1966) 239–256.
- C.J. van Oss, R.J. Good, and M.K. Chaudhury, Langmuir. 4 (1988) 884–891.
- E. Cremer and H. Huber, in Gas Chromatography., ed. N. Brenner, et al., Academic Press, New York (1962) p 169.
- P.P. Yla-Maihaniemi, J.Y.Y. Heng, F. Thielmann, and D.R. Williams, Langmuir. 24 (2008) 9551–9557.
- F. Thielmann, D.J. Burnett, and J.Y.Y. Heng, Drug Development and Industrial Pharmacy. 33 (2007) 1240–1253.
- J. van Deemter, F.J. Zuiderweg, and A. Klinkenberg, Chemical Engineering Science. 5 (1965) 271.
- G. Buckton, A. Ambarkhane, and K. Pincott, Pharmaceutical Research. 21 (2004) 1554–1557.
- 14 D. Benczedi, D, I. Tomka, and F. Escher, Macromolecules. 31 (1998) 3055.
- G. DiPaola and J.E. Guillet, Macromolecules. 11 (1978) 228.
- K. Adamska and A. Voelkel, International Journal of Pharmaceutics. 304 (2005) 11–17.
- G.J. Price, K.S. Siow, and J.E. Guillet, Macromolecules. 22 (1989) 3116–3119.
- A. Ziani, R. Xu, H.P. Schreiber, and T. Kobayashi, Journal of Coatings Technology. 71 (1999) 53–60.
- A. Lavole and J.E. Guillet, Macromolecules. 2 (1969) 443.
- F. Thielmann and D.R. Williams, Deutsche Lebensmittel-Rundschau. 96 (2000) 255–257.
- H.H.Y. Tong, B.Y. Shekunov, P. York, and A.H.L Chow, Pharmaceutical Research. 19 (2002) 640–648.
- J.Y.Y. Heng, F. Thielmann, and D.R. Williams, Pharmaceutical Research, 23 (2006) 1918–1927.
- J. Feeley, P. York, B. Sumby, and H. Dicks, International Journal of Pharmaceutics. 172 (1998) 89–96.
- N. Ahfat, G. Buckton, R. Burrows, and M. Ticehurst, European Journal of Pharmaceutical Sciences. 9 (2000) 271–276.
- E. Cantergiani and D. Benczedi, Journal of Chromatography A. 969 (2002) 103–110.
- J.Y.Y. Heng, D.F. Pearse, F. Thielmann, T. Lampke, and A. Bismarck, Composite Interfaces. 14 (2007) 581–604.
- K. Tsutsumi and T. Ohsuga, Colloid and Polymer Science. 268 (1990) 38–44.
- L. Lavielle and J. Schultz, Langmuir. 7 (1991) 978–981.
- K. Batko and A. Voelkel, Journal of Colloid and Interface Science. 315 (2007) 768–771.
- R. Menzel, A. Lee, A. Bismarck, and M.S.P. Shaffer, Langmuir. (2009) in press.
- Gamelas, J.; Ferraz, E.; Rocha, F. (2014). "An insight into the surface properties of calcined kaolinitic clays: the grinding effect". Colloids and Surfaces A: Physicochemical and Engineering Aspects. 455: 49–57. doi:10.1016/j.colsurfa.2014.04.038.
- J. Borch, Journal of Adhesion Science and Technology. 5 (1991) 523–541.
- R.H. Mills, D.J. Gardner, and R. Wimmer. Journal of Applied Polymer Science. 110 (2008) 3880–3888.
- Q. Zhou and K.R. Cadwallader. Journal of Agricultural and Food Chemistry. 54 (2006) 1838–1843.