Variant Creutzfeldt–Jakob disease
| Variant Creutzfeldt–Jakob disease | |
|---|---|
| Synonyms | New variant Creutzfeldt–Jakob disease (nvCJD) |
 | |
| Biopsy of the tonsil in variant CJD. Prion protein immunostaining. | |
| Specialty | Neurology |
| Symptoms | Psychiatric problems, behavioral changes, painful sensations[1] |
| Usual onset | Less than 30 years old[2] |
| Causes | Eating beef from animals with bovine spongiform encephalopathy[2][3] |
| Diagnostic method | Brain biopsy[2] |
| Treatment | Supportive care[4] |
| Prognosis | ~13 month life expectancy[1] |
| Frequency | Less than 250 reported cases as of 2012[5] |
Variant Creutzfeldt–Jakob disease (vCJD) is a type of brain disease within the transmissible spongiform encephalopathy family.[5] Symptoms include psychiatric problems, behavioral changes, and painful sensations.[1] The length of time between exposure and the development of symptoms is unclear, but is believed to be years.[2] Average life expectancy following the onset of symptoms is 13 months.[1]
It is caused by prions, which are mis-folded proteins.[6] Spread is believed to be primarily due to eating bovine spongiform encephalopathy (BSE)-infected beef.[5][6] Infection is also believed to require a specific genetic susceptibility.[3][5] Spread may potentially also occur via blood products or contaminated surgical equipment.[7] Diagnosis is by brain biopsy but can be suspected based on certain other criteria.[2] It is a different from classic Creutzfeldt–Jakob disease, though both are due to prions.[6]
Treatment involves supportive care.[4] As of 2012 about 170 cases of vCJD have been recorded in the United Kingdom, and 50 cases in the rest of the world.[5] The disease has become less common since 2000.[5] The typical age of onset is less than 30 years old.[2] It was first identified in 1996 by the National CJD Surveillance Unit in Edinburgh, Scotland.[5]
Cause
Blood products
As of 2018 evidence suggest that while there may be prions in the blood of individuals with vCJD, this is not the case in individuals with sporadic CJD.[8]
In 2004, a report published in the medical journal The Lancet showed that vCJD can be transmitted by blood transfusions.[9] The finding alarmed healthcare officials because a large epidemic of the disease could result in the near future. A blood test for vCJD infection is possible[10] but is not yet available for screening blood donations. Significant restrictions exist to protect the blood supply. The UK government banned anyone who had received a blood transfusion since January 1980 from donating blood.[11] Since 1999 there has been a ban in the UK for using UK blood to manufacture fractional products such as albumin.[12] Whilst these restrictions may go some way to preventing a self-sustaining epidemic of secondary infections the number of infected blood donations is unknown and could be considerable as a study by the Health Protection Agency show around 1 in 2000 people in the UK shows signs of abnormal prion accumulation.[13] In June 2013 the government was warned that deaths—then at 176—could rise five-fold through blood transfusions.[14]
On May 28, 2002, the United States Food and Drug Administration instituted a policy that excludes from donation anyone having spent at least six months in certain European countries (or three months in the United Kingdom) from 1980 to 1996. Given the large number of U.S. military personnel and their dependents residing in Europe, it was expected that over 7% of donors would be deferred due to the policy. Later changes to this policy have relaxed the restriction to a cumulative total of five years or more of civilian travel in European countries (six months or more if military). The three-month restriction on travel to the UK, however, has not been changed.[15]
In New Zealand, the New Zealand Blood Service (NZBS) in 2000 introduced measures to preclude permanently donors having resided in the United Kingdom (including the Isle of Man and the Channel Islands) for a total of six months or more between January 1980 and December 1996. The measure resulted in ten percent of New Zealand's active blood donors at the time becoming ineligible to donate blood. In 2003, the NZBS further extended restrictions to permanently preclude donors having received a blood transfusion in the United Kingdom since January 1980, and in April 2006, restrictions were further extended to include the Republic of Ireland and France.[16]
Similar regulations are in place in France and in Germany, where anyone having spent six months or more living in the UK between January 1980 and December 1996 is permanently banned from donating blood.[17]
In Canada, individuals are not eligible to donate blood or plasma if they have spent a cumulative total of three months or more in the United Kingdom (UK) or France from January 1, 1980 to December 31, 1996. They are also ineligible if they have spent a cumulative total of five years or more in Western Europe outside the U.K. or France since January 1, 1980 through December 31, 2007 or spent a cumulative total of six months or more in Saudi Arabia from January 1, 1980 through December 31, 1996[18] or if they have had a blood transfusion in the U.K., France or Western Europe since 1980.[19]
In Poland, anyone having spent cumulatively six months or longer between 1 January 1980 and 31 December 1996 in the UK, Ireland, or France is permanently barred from donating.[20]
In the Czech Republic, anyone having spent more than six months in the UK or France between the years 1980 and 1996 or received transfusion in the UK after the year 1980 is not allowed to donate blood.[21]
Sperm donation
In the U.S., the FDA has banned import of any donor sperm, motivated by a risk of variant Creutzfeldt-Jakob disease, inhibiting the once popular[22] import of Scandinavian sperm. Despite this the scientific consensus is that the risk is negligible, as there is no evidence Creutzfeldt–Jakob is sexually transmitted.[23][24][25]
Mechanism
Despite the consumption of contaminated beef in the UK being high, vCJD has infected a small number of people. One explanation for this can be found in the genetics of patients with the disease. The human PRNP protein which is subverted in prion disease can occur with either methionine or valine at amino acid 129, without any apparent difference in normal function. Of the overall Caucasian population, about 40% have two methionine-containing alleles, 10% have two valine-containing alleles, and the other 50% are heterozygous at this position. Only a single person with vCJD tested was found to be heterozygous; most of those affected had two copies of the methionine-containing form. It is not yet known whether those unaffected are actually immune or only have a longer incubation period until symptoms appear.[26][27]
Diagnosis
Definitive
Examination of brain tissue is required to confirm a diagnosis of variant CJD.[28] The following confirmatory features should be present:[28]
Suspected
- Current age or age at death less than 55 years (a brain autopsy is recommended, however, for all physician-diagnosed CJD cases).[28]
- Psychiatric symptoms at illness onset and/or persistent painful sensory symptoms (frank pain and/or dysesthesia).[28]
- Dementia, and development ≥4 months after illness onset of at least two of the following five neurologic signs: poor coordination, myoclonus, chorea, hyperreflexia, or visual signs. (If persistent painful sensory symptoms exist, ≥4 months delay in the development of the neurologic signs is not required).[28]
- A normal or an abnormal EEG, but not the diagnostic EEG changes often seen in classic CJD.[28]
- Duration of illness of over 6 months.[28]
- Routine investigations do not suggest an alternative, non-CJD diagnosis.[28]
- No history of receipt of cadaveric human pituitary growth hormone or a dura mater graft.[28]
- No history of CJD in a first degree relative or prion protein gene mutation in the person.[28]
Society and culture
United Kingdom
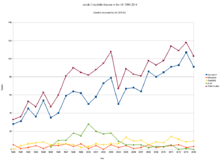
Researchers believe one in 2,000 people in the UK is a carrier of the disease linked to eating contaminated beef (vCJD).[29] The survey provides the most robust prevalence measure to date—and identifies abnormal prion protein across a wider age group than found previously and in all genotypes, indicating "infection" may be relatively common. This new study examined over 32,000 anonymous appendix samples. Of these, 16 samples were positive for abnormal prion protein, indicating an overall prevalence of 493 per million population, or one in 2,000 people are likely to be carriers. No difference was seen in different birth cohorts (1941–60 and 1961–85), in both sexes, and there was no apparent difference in abnormal prion prevalence in three broad geographical areas. Genetic testing of the 16 positive samples revealed a higher proportion of valine homozygous (VV) genotype on the codon 129 of the gene encoding the prion protein (PRNP) compared with the general UK population. This also differs from the 177 patients with vCJD, all of whom to date have been methionine homozygous (MM) genotype. The concern is that individuals with this VV genotype may be susceptible to developing the condition over longer incubation periods.[30]
References
- 1 2 3 4 "Clinical and Pathologic Characteristics | Variant Creutzfeldt-Jakob Disease, Classic (CJD)". CDC. 10 February 2015. Retrieved 22 January 2018.
- 1 2 3 4 5 6 "Classic CJD versus Variant CJD". CDC. 11 February 2015. Retrieved 23 January 2018.
- 1 2 Ironside, JW (Jul 2010). "Variant Creutzfeldt–Jakob disease". Haemophilia. 16 Suppl 5: 175–80. doi:10.1111/j.1365-2516.2010.02317.x. PMID 20590878.
- 1 2 "Treatment Variant Creutzfeldt-Jakob Disease". CDC. 10 February 2015. Retrieved 23 January 2018.
- 1 2 3 4 5 6 7 Ironside, JW (2012). "Variant Creutzfeldt–Jakob disease: an update". Folia Neuropathologica. 50 (1): 50–6. PMID 22505363.
- 1 2 3 "About vCJD". CDC. 10 February 2015. Retrieved 22 January 2018.
- ↑ Ferri, Fred F. (2017). Ferri's Clinical Advisor 2018 E-Book: 5 Books in 1. Elsevier Health Sciences. p. 343. ISBN 9780323529570.
- ↑ "Creutzfeldt-Jakob Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". NINDS. March 2003. Archived from the original on 4 July 2017. Retrieved 16 July 2017.
- ↑ Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW (2004). "Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient". The Lancet. 364 (9433): 527–9. doi:10.1016/S0140-6736(04)16811-6. PMID 15302196.
- ↑ Edgeworth, JA; Farmer, M; Sicilia, A; Tavares, P; Beck, J; Campbell, T; Lowe, J; Mead, S; Rudge, P; Collinge, J; Jackson, GS (Feb 5, 2011). "Detection of prion infection in variant Creutzfeldt–Jakob disease: a blood-based assay". The Lancet. 377 (9764): 487–93. doi:10.1016/S0140-6736(10)62308-2. PMID 21295339.
- ↑ "Variant CJD and blood donation" (PDF). National Blood Service. August 2004. Archived from the original (PDF) on October 11, 2007. Retrieved 2009-06-20.
- ↑ Regan F, Taylor C (July 2002). "Blood transfusion medicine". BMJ (Clinical Research Ed.). 325 (7356): 143–7. doi:10.1136/bmj.325.7356.143. PMC 1123672. PMID 12130612.
- ↑ HPA Press Office (August 10, 2012). "Summary results of the second national survey of abnormal prion prevalence in archived appendix specimens". Archived from the original on March 25, 2013.
- ↑ Rowena Mason (April 28, 2013). "Mad cow infected blood 'to kill 1,000'". Daily Telegraph. London. Archived from the original on July 3, 2013. Retrieved July 2, 2013.
- ↑ "In-Depth Discussion of Variant Creutzfeld–Jacob Disease and Blood Donation". American Red Cross. Archived from the original on 2007-12-30. Retrieved 2009-06-20.
- ↑ "CJD (Creutzfeldt–Jakob Disease) - Information for blood donors" (PDF). New Zealand Blood Service. Archived (PDF) from the original on 10 April 2017. Retrieved 31 May 2017.
- ↑ "Permanent exclusion criteria" (in German). Blutspendedienst Hamburg. Archived from the original on 9 August 2016. Retrieved 2009-06-20. English via Google Translate
- ↑ "Canada restricts blood donors from Saudi Arabia". ctv news. Archived from the original on 5 May 2016. Retrieved 14 April 2016.
- ↑ "Travel restrication". Canadian Blood Services. Archived from the original on 14 April 2016. Retrieved 14 April 2016.
- ↑ "Permanent exclusion criteria / Dyskwalifikacja stała" (in Polish). RCKiK Warszawa. Archived from the original on August 30, 2009. Retrieved 2010-03-03.
- ↑ "Blood donor guidance / Poučení dárce krve" (in Czech). Fakultní nemocnice Královské Vinohrady. Archived from the original on 2011-07-18. Retrieved 2010-03-20.
- ↑ Stein, Rob (13 August 2008). "Mad Cow Rules Hit Sperm Banks' Patrons". washingtonpost.com. The Washington Post Company. Archived from the original on 26 April 2012. Retrieved 4 October 2008.
- ↑ Kotler, Steven (27 September 2007). "The God of Sperm". LA Weekly. Archived from the original on 6 July 2009. Retrieved 20 June 2009.
- ↑ "Is there a real risk of transmitting variant Creutzfeldt-Jakob disease by donor sperm insemination?". Reproductive BioMedicine Online. 2006. doi:10.1016/S1472-6483(10)61024-3. Retrieved 2017-01-07.
- ↑ Lapidos, Juliet (26 September 2007). "Is Mad Cow an STD?". Slate. Archived from the original on 8 January 2017. Retrieved 7 January 2017.
- ↑ Saba, R; Booth, SA (2013). "The genetics of susceptibility to variant Creutzfeldt–Jakob disease". Public Health Genomics. 16 (1–2): 17–24. doi:10.1159/000345203. PMID 23548713.
- ↑ Sikorska, B; Liberski, PP (2012). "Human prion diseases: from Kuru to variant Creutzfeldt–Jakob disease". Sub-cellular biochemistry. 65: 457–96. doi:10.1007/978-94-007-5416-4_17. PMID 23225013.
- 1 2 3 4 5 6 7 8 9 10 11 12 "Diagnostic Criteria | Variant Creutzfeldt-Jakob Disease, Classic (CJD)". CDC. 10 February 2015. Retrieved 23 January 2018.
- ↑ "Estimate doubled for vCJD carriers in UK". BBC News. 2013-10-15. Archived from the original on 2014-02-10.
- ↑ Gill, Noel (2013). "Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey". British Medical Journal. 347: f5675. doi:10.1136/bmj.f5675. PMC 3805509. PMID 24129059. Retrieved 27 December 2013.