Single cell sequencing
Single cell sequencing examines the sequence information from individual cells with optimized next generation sequencing (NGS) technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment.[1]
Background
A typical human cell consists of about 2 x 3.3 billion base pairs of DNA and 600 million bases of mRNA. Usually a mix of millions of cells are used in sequencing the DNA or RNA using traditional methods like Sanger sequencing or Illumina sequencing. By using deep sequencing of DNA and RNA from a single cell, cellular functions can be investigated extensively.[1] Like typical NGS experiments, the protocols of single cell sequencing generally contain the following steps: isolation of a single cell, nucleic acid extraction and amplification, sequencing library preparation, sequencing and bioinformatic data analysis. It is more challenging to perform single cell sequencing in comparison with sequencing from cells in bulk. The minimal amount of starting materials from a single cell make degradation, sample loss and contamination exert pronounced effects on quality of sequencing data. In addition, due to the picogram level of the amount of nucleic acids used,[2] heavy amplification is often needed during sample preparation of single cell sequencing, resulting in the uneven coverage, noise and inaccurate quantification of sequencing data.
Recent technical improvements make single cell sequencing a promising tool for approaching a set of seemingly inaccessible problems. For example, heterogeneous samples, rare cell types, cell lineage relationships, mosaicism of somatic tissues, analyses of microbes that cannot be cultured, and disease evolution can all be elucidated through single cell sequencing.[3] Single cell sequencing was selected as the method of the year 2013 by Nature Publishing Group.[4]
Single cell genome (DNA) sequencing
Single cell DNA genome sequencing involves isolating a single cell, performing whole-genome-amplification (WGA), constructing sequencing libraries and then sequencing the DNA using a next-generation sequencer (ex. Ion Torrent, Illumina). A genome constructed in this fashion is commonly referred to as a single amplified genome or SAG. It can be used in microbiome studies, in order to obtain genomic data from uncultured microorganisms. In addition, it can be united with high throughput cell sorting of microorganisms and cancer. One popular method used for single cell genome sequencing is multiple displacement amplification and this enables research into various areas such as microbial genetics, ecology and infectious diseases. Furthermore, data obtained from microorganisms might establish processes for culturing in the future.[5] Some of the genome assembly tools that can be used in single cell genome sequencing include: SPAdes, IDBA-UD, Cortex and HyDA.[6]
Method
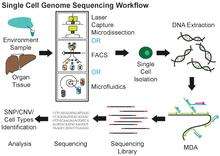
Multiple displacement amplification (MDA) is a widely used technique, enabling amplifying femtograms of DNA from bacterium to micrograms for the use of sequencing. Reagents required for MDA reactions include: random primers and DNA polymerase from bacteriophage phi29. In 30 degree isothermal reaction, DNA is amplified with included reagents. As the polymerases manufacture new strands, a strand displacement reaction takes place, synthesizing multiple copies from each template DNA. At the same time, the strands that were extended antecedently will be displaced. MDA products result in a length of about 12 kb and ranges up to around 100 kb, enabling its use in DNA sequencing.[5] In 2017, a major improvement to this technique, called WGA-X, was introduced by taking advantage of a thermostable mutant of the phi29 polymerase, leading to better genome recovery from individual cells, in particular those with high G+C content.[7] Other methods include MALBAC.[8]
Limitations
MDA of individual cell genomes results in highly uneven genome coverage, i.e. relative overrepresentation and underrepresentation of various regions of the template, leading to loss of some sequences. There are two components to this process: a) stochastic over- and under-amplification of random regions; and b) systematic bias against high %GC regions. The stochastic component may be addressed by pooling single-cell MDA reactions from the same cell type, by employing fluorescent in situ hybridization (FISH) and/or post-sequencing confirmation.[5] The bias of MDA against high %GC regions can be addressed by using thermostable polymerases, such as in the process called WGA-X.[7]
Single-nucleotide polymorphisms (SNPs), which are a big part of genetic variation in the human genome, and copy number variation (CNV), pose problems in single cell sequencing, as well as the limited amount of DNA extracted from a single cell. Due to scant amounts of DNA, accurate analysis of DNA poses problems even after amplification since coverage is low and susceptible to errors. With MDA, average genome coverage is less than 80% and SNPs that are not covered by sequencing reads will be opted out. In addition, MDA shows a high ratio of allele dropout, not detecting alleles from heterozygous samples. Various SNP algorithms are currently in use but none are specific to single cell sequencing. MDA with CNV also poses the problem of identifying false CNVs that conceal the real CNVs. To solve this, when patterns can be generated from false CNVs, algorithms can detect and eradicate this noise to produce true variants.[8]
Applications
Microbiomes are among the main targets of single cell genomics due to the difficulty of culturing the majority of microorganisms in most environments. Single cell genomics is a powerful way to obtain microbial genome sequences without cultivation. This approach has been widely applied on marine, soil, subsurface, organismal, and other types of microbiomes in order to address a wide array of questions related to microbial ecology, evolution, public health and biotechnology potential.[9][10][11][12][13][14][15][16][17][18][7][19]
Cancer sequencing is also an emerging application of scDNAseq. Fresh or frozen tumors may be analyzed and categorized with respect to SCNAs, SNVs, and rearrangements quite well using whole genome DNAS approaches [20] Cancer scDNAseq is particularly useful for examining the depth of complexity and compound mutations present in amplified therapeutic targets such as receptor tyrosine kinase genes (EGFR, PDGFRA etc.) where conventional population-level approaches of the bulk tumor are not able to resolve the co-occurrence patterns of these mutations within single cells of the tumor. Such overlap may provide redundancy of pathway activation and tumor cell resistance.
Single cell DNA methylome sequencing
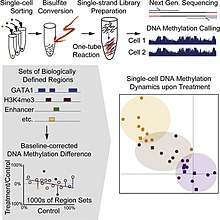
Single cell DNA methylome sequencing quantifies DNA methylation. This is similar to single cell genome sequencing, but with the addition of a bisulfite treatment before sequencing. Forms include whole genome bisulfite sequencing,[21][22] and reduced representation bisulfite sequencing [23][24]

Single-cell RNA sequencing (scRNA-seq)
Current methods for quantifying molecular states of cells, from microarray to standard RNA-seq analysis, mostly depend on estimating the mean value from millions of cells by averaging the signal of individual cells. Given the heterogeneity of cell population, measurement of the mean values of signals overlooks the internal interactions and differences within a cell population that may be crucial for maintaining normal tissue functions and facilitating disease progression. Thus the cell-averaging experiments provide only partial information of the molecular state of the system.[25][26]
Single-cell RNA sequencing (scRNA-seq) provides the expression profile of individual cells. Through gene clustering analyses, rare cell types within a cell population can be identified, thereby making characterization of the subpopulation structure of a heterogeneous cell population possible. While tumor heterogeneity can be attributed to accumulated mutations, even genetically identical cells, under the same environment, display high variability of gene and protein expression levels.[27] However, RNA with low copy number, which may exert important functions in the cells, is usually undetectable or regarded as noise in traditional cell-averaging methodsNeeds citation. Single-cell RNA sequencing on a large number of single cells can identify such uncommon RNA and also reveal the copy-number distribution of the whole mRNA population in individual cells.[26][28]
Experimental procedures
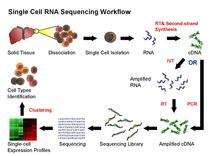
Despite the advances in sequencing technologies, it is currently not possible to sequence RNA directly from a single cell. Thus, in the current scRNA-seq protocols, RNA still needs to be converted to cDNA for sequencing. Principally, the current scRNA-seq methods contain the following steps: isolation of single cell and RNA, reverse transcription (RT), amplification, library generation and sequencing.
The ideal scRNA-seq preserves and accurately quantifies the initial relative abundance of mRNA in a cell, covers the entire transcript lengths with equal representation at each position, and retains strand information.[28] However, a variety of noise and bias may be introduced in various steps of scRNA-seq protocol. For example, the step of reverse transcription is critical as the efficiency of the RT reaction determines the percentage of a cell’s RNA population that is eventually analyzed by the sequencer. The processivity of reverse transcriptases and the priming strategies used will affect full-length cDNA production and the generation of libraries biased toward 3’ or 5' end of genes.
In the amplification step, either PCR or in vitro transcription (IVT) is currently used to amplify cDNA. One of the advantages of PCR-based methods is able to generate full-length cDNA. However, different PCR efficiency on particular sequences (for instance, GC content and snapback structure) will also be exponentially amplified, producing libraries with uneven coverage. On the other hand, while libraries generated by IVT can avoid PCR-induced sequence bias, specific sequences may be transcribed inefficiently, thus causing sequence drop-out or generating incomplete sequences.[1][25] Several scRNA-seq protocols have been published: Tang et al.,[29] STRT,[30] SMART-seq,[31] CEL-seq,[32] RAGE-seq, [33] and Quartz-seq.[34] These protocols differ in terms of strategies for reverse transcription, cDNA synthesis and amplification, and the possibility to accommodate sequence-specific barcodes (i.e. UMIs) or the ability to process pooled samples.[35]
Applications
The number of circulating tumor cells (CTC) in peripheral blood of cancer patients has been shown to correlate to prognosis.[36] However, it is challenging to enumerate and characterize the isolated CTCs as they are often contaminated with a large number of leukocytes and erythrocytes. Single cell RNA-seq could be applied to differentiate cancer cells from normal blood cells and obtain the expression profiles of tumor cells at the same time. Similarly, single cell RNA-seq can also be used to analyze rare cell types in early human embryo and adult stem cells, both of which exist transiently and difficult to be characterized with current technologies. Finally, single cell analysis can be applied to the study of infectious diseases.[37]
Considerations
Isolation of single cells
There are several ways to isolate individual cells prior to whole genome amplification and sequencing. Fluorescence-activated cell sorting (FACS) is the most widely used approach and is employed by several high-throughput core facilities, such as Bigelow Laboratory Single Cell Genomics Center.[38] Individual cells can also be collected by micromanipulation, for example by serial dilution or by using a patch pipette or nanotube to harvest a single cell.[39][40] The advantages of micromanipulation are ease and low cost, but they are laborious and susceptible to misidentification of cell types under microscope. Laser-capture microdissection (LCM) can also be used for collecting single cells. Although LCM preserves the knowledge of the spatial location of a sampled cell within a tissue, it is hard to capture a whole single cell without also collecting the materials from neighboring cells.[25][41][42] High-throughput methods for single cell isolation also include microfluidics. Both of FACS and microfluidics are accurate, automatic and capable of isolating unbiased samples. However, both methods require detaching cells from their microenvironments first, thereby causing perturbation to the transcriptional profiles in RNA expression analysis.[43][44]
Number of cells to be analyzed
scRNA-Seq
Generally speaking, for a typical bulk cell RNA-sequencing (RNA-seq) experiment, ten million reads are generated and a gene with higher than the threshold of 50 reads per kb per million reads (RPKM) is considered expressed. For a gene that is 1kb long, this corresponds to 500 reads and a minimum coefficient of variation (CV) of 4% under the assumption of the Poisson distribution. For a typical mammalian cell containing 200,000 mRNA, sequencing data from at least 50 single cells need to be pooled in order to achieve this minimum CV value. However, due to the efficiency of reverse transcription and other noise introduced in the experiments, more cells are required for accurate expression analyses and cell type identification.[25]
See also
References
- 1 2 3 Eberwine J, Sul JY, Bartfai T, Kim J (January 2014). "The promise of single-cell sequencing". Nat. Methods. 11 (1): 25–27. doi:10.1038/nmeth.2769. PMID 24524134.
- ↑ Shintaku H, Nishikii H, Marshall LA, Kotera H, Santiago JG (2014). "On-Chip Separation and Analysis of RNA and DNA from Single Cells". Anal. Chem. 86 (4): 1953–7. doi:10.1021/ac4040218. PMID 24499009.
- ↑ Nawy, T. (2013). "Single-cell sequencing". Nat Methods. 11 (1): 18. doi:10.1038/nmeth.2771. PMID 24524131.
- ↑ "Method of the Year 2013". Nat Methods. 11 (1). 2014. doi:10.1038/nmeth.2801.
- 1 2 3 "Lasken, RS. (2007). "Single-cell Genomic Sequencing using Multiple Displacement Amplification". Curr Opin Microbiol. 10 (5): 510–16. doi:10.1016/j.mib.2007.08.005. PMID 17923430. "
- ↑ Taghavi, Z.; Movahedi NS; Draghici S; Chitsaz H. (2013). "Distilled single-cell genome sequencing and de novo assembly for sparse microbial communities". Bioinformatics. 29 (19): 2395–2401. arXiv:1305.0062. doi:10.1093/bioinformatics/btt420.
- 1 2 3 Stepanauskas, Ramunas; Fergusson, Elizabeth A.; Brown, Joseph; Poulton, Nicole J.; Tupper, Ben; Labonté, Jessica M.; Becraft, Eric D.; Brown, Julia M.; Pachiadaki, Maria G. (2017-07-20). "Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles". Nature Communications. 8 (1). Bibcode:2017NatCo...8...84S. doi:10.1038/s41467-017-00128-z. ISSN 2041-1723.
- 1 2 "Ning, L.; Liu G; Li G; Hou Y; Tong Y; He J. (2014). "Current Challenges in the Bioinformatics of Single Cell Genomics". Front Oncol. 4 (7). doi:10.3389/fonc.2014.00007. PMC 3902584. PMID 24478987. "
- ↑ Blainey, PC.; Quake SR. (2014). "Dissecting genomic diversity, one cell at a time". Nat Methods. 11 (1): 19–21. doi:10.1038/nmeth.2783. PMC 3947563. PMID 24524132.
- ↑ Zhang; Martiny, AC; Reppas, NB; Barry, KW; Malek, J; Chisholm, SW; Church, GM (2006). "Sequencing genomes from single cells by polymerase cloning". Nature Biotechnology. 24 (6): 680–6. doi:10.1038/nbt1214. PMID 16732271.
- ↑ Stepanauskas, Ramunas; Sieracki, Michael E. (2007-05-22). "Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time". Proceedings of the National Academy of Sciences. 104 (21): 9052–9057. Bibcode:2007PNAS..104.9052S. doi:10.1073/pnas.0700496104. ISSN 0027-8424. PMC 1885626. PMID 17502618.
- ↑ Yoon, Hwan Su; Price, Dana C.; Stepanauskas, Ramunas; Rajah, Veeran D.; Sieracki, Michael E.; Wilson, William H.; Yang, Eun Chan; Duffy, Siobain; Bhattacharya, Debashish (2011-05-06). "Single-Cell Genomics Reveals Organismal Interactions in Uncultivated Marine Protists". Science. 332 (6030): 714–717. Bibcode:2011Sci...332..714Y. doi:10.1126/science.1203163. ISSN 0036-8075. PMID 21551060.
- ↑ Swan, Brandon K.; Martinez-Garcia, Manuel; Preston, Christina M.; Sczyrba, Alexander; Woyke, Tanja; Lamy, Dominique; Reinthaler, Thomas; Poulton, Nicole J.; Masland, E. Dashiell P. (2011-09-02). "Potential for Chemolithoautotrophy Among Ubiquitous Bacteria Lineages in the Dark Ocean". Science. 333 (6047): 1296–1300. Bibcode:2011Sci...333.1296S. doi:10.1126/science.1203690. ISSN 0036-8075. PMID 21885783.
- ↑ Woyke, Tanja; Xie, Gary; Copeland, Alex; González, José M.; Han, Cliff; Kiss, Hajnalka; Saw, Jimmy H.; Senin, Pavel; Yang, Chi (2009-04-23). "Assembling the Marine Metagenome, One Cell at a Time". PLOS ONE. 4 (4): e5299. Bibcode:2009PLoSO...4.5299W. doi:10.1371/journal.pone.0005299. ISSN 1932-6203. PMC 2668756. PMID 19390573.
- ↑ Swan, Brandon K.; Tupper, Ben; Sczyrba, Alexander; Lauro, Federico M.; Martinez-Garcia, Manuel; González, José M.; Luo, Haiwei; Wright, Jody J.; Landry, Zachary C. (2013-07-09). "Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean". Proceedings of the National Academy of Sciences. 110 (28): 11463–11468. Bibcode:2013PNAS..11011463S. doi:10.1073/pnas.1304246110. ISSN 0027-8424. PMID 23801761.
- ↑ Rinke, Christian; Schwientek, Patrick; Sczyrba, Alexander; Ivanova, Natalia N.; Anderson, Iain J.; Cheng, Jan-Fang; Darling, Aaron; Malfatti, Stephanie; Swan, Brandon K. (2013-07). "Insights into the phylogeny and coding potential of microbial dark matter". Nature. 499 (7459): 431–437. Bibcode:2013Natur.499..431R. doi:10.1038/nature12352. ISSN 0028-0836. Check date values in:
|date=(help) - ↑ Kashtan, Nadav; Roggensack, Sara E.; Rodrigue, Sébastien; Thompson, Jessie W.; Biller, Steven J.; Coe, Allison; Ding, Huiming; Marttinen, Pekka; Malmstrom, Rex R. (2014-04-25). "Single-Cell Genomics Reveals Hundreds of Coexisting Subpopulations in Wild Prochlorococcus". Science. 344 (6182): 416–420. Bibcode:2014Sci...344..416K. doi:10.1126/science.1248575. ISSN 0036-8075. PMID 24763590.
- ↑ Wilson, William H; Gilg, Ilana C; Moniruzzaman, Mohammad; Field, Erin K; Koren, Sergey; LeCleir, Gary R; Martínez Martínez, Joaquín; Poulton, Nicole J; Swan, Brandon K (2017-05-12). "Genomic exploration of individual giant ocean viruses". The ISME Journal. 11 (8): 1736–1745. doi:10.1038/ismej.2017.61. ISSN 1751-7362.
- ↑ Pachiadaki, Maria G.; Sintes, Eva; Bergauer, Kristin; Brown, Julia M.; Record, Nicholas R.; Swan, Brandon K.; Mathyer, Mary Elizabeth; Hallam, Steven J.; Lopez-Garcia, Purificacion (2017-11-24). "Major role of nitrite-oxidizing bacteria in dark ocean carbon fixation". Science. 358 (6366): 1046–1051. Bibcode:2017Sci...358.1046P. doi:10.1126/science.aan8260. ISSN 0036-8075. PMID 29170234.
- ↑ (Francis J, Zheng CZ, Maire C et al. Cancer Discovery 2014).
- 1 2 Farlik, M; Sheffield, NC; Nuzzo, A; Datlinger, P; Schönegger, A; Klughammer, J; Bock, C (3 March 2015). "Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics". Cell Reports. 10 (8): 1386–97. doi:10.1016/j.celrep.2015.02.001. PMC 4542311. PMID 25732828.
- ↑ Smallwood, SA; Lee, HJ; Angermueller, C; Krueger, F; Saadeh, H; Peat, J; Andrews, SR; Stegle, O; Reik, W; Kelsey, G (August 2014). "Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity". Nature Methods. 11 (8): 817–20. doi:10.1038/nmeth.3035. PMC 4117646. PMID 25042786.
- ↑ Guo, H; Zhu, P; Wu, X; Li, X; Wen, L; Tang, F (December 2013). "Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing". Genome Research. 23 (12): 2126–35. doi:10.1101/gr.161679.113. PMC 3847781. PMID 24179143.
- ↑ Guo, H; Zhu, P; Guo, F; Li, X; Wu, X; Fan, X; Wen, L; Tang, F (May 2015). "Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing". Nature Protocols. 10 (5): 645–59. doi:10.1038/nprot.2015.039. PMID 25837417.
- 1 2 3 4 "Shapiro, E.; Biezuner T; Linnarsson S. (2013). "Single-cell sequencing-based technologies will revolutionize whole-organism science". Nat Rev Genet. 14 (9): 618–30. doi:10.1038/nrg3542. PMID 23897237. "
- 1 2 Kolodziejczyk, Aleksandra A.; Kim, Jong Kyoung; Svensson, Valentine; Marioni, John C.; Teichmann, Sarah A. (May 2015). "The Technology and Biology of Single-Cell RNA Sequencing". Molecular Cell. 58 (4): 610–620. doi:10.1016/j.molcel.2015.04.005.
- ↑ Munsky B, Neuert G, van Oudenaarden A (2012). "Using gene expression noise to understand gene regulation". Science. 336 (6078): 183–7. Bibcode:2012Sci...336..183M. doi:10.1126/science.1216379. PMC 3358231. PMID 22499939.
- 1 2 "Hebenstreit, D. (2012). "Methods, Challenges and Potentials of Single Cell RNA-seq". Biology. 1 (3): 658–667. doi:10.3390/biology1030658. "
- ↑ Tang, F.; Barbacioru C; Wang Y; et al. (2009). "mRNA-Seq whole-transcriptome analysis of a single cell". Nat Methods. 6 (5): 377–82. doi:10.1038/NMETH.1315. PMID 19349980.
- ↑ Islam, S.; Kjällquist U; Moliner A; et al. (2011). "Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq". Genome Res. 21 (7): 1160–7. doi:10.1101/gr.110882.110. PMC 3129258. PMID 21543516.
- ↑ Ramsköld, D.; Luo S; Wang Y-C; et al. (2012). "Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells". Nat. Biotechnol. 30 (8): 777–82. doi:10.1038/nbt.2282. PMC 3467340. PMID 22820318.
- ↑ Hashimshony, T.; Wagner F; Sher N; Yanai I. (2012). "CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification". Cell Rep. 2 (3): 666–73. doi:10.1016/j.celrep.2012.08.003. PMID 22939981.
- ↑ Singh, M.; Ghamdan A.; Smith M. A.; Swarbrick A. (2018). "High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes". BioRxiv. doi:10.1101/424945.
- ↑ Sasagawa, Y.; Nikaido I; Hayashi T; et al. (2013). "Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity". Genome Biol. 14 (4): R31. doi:10.1186/gb-2013-14-4-r31. PMC 4054835. PMID 23594475.
- ↑ Dal Molin, A.; Di Camillo B (2018). "How to design a single-cell RNA-sequencing experiment: pitfalls, challenges and perspectives". Briefings in Bioinformatics: bby007. doi:10.1093/bib/bby007. PMID 29394315.
- ↑ Olmos, D.; Arkenau H-T; Ang JE; et al. (2009). "Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): a single-centre experience". Ann. Oncol. 20 (1): 27–33. doi:10.1093/annonc/mdn544. PMID 18695026.
- ↑ Avraham, R; Haseley, N; Brown, D; Penaranda, C; Jijon, HB; Trombetta, JJ; Satija, R; Shalek, AK; Xavier, RJ; Regev, A; Hung, DT (10 September 2015). "Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses". Cell. 162 (6): 1309–21. doi:10.1016/j.cell.2015.08.027. PMC 4578813. PMID 26343579.
- ↑ "Single Cell Genomics Center :: Home". scgc.bigelow.org. Retrieved 2018-05-25.
- ↑ Zong, C; Lu S; Chapman AR; Xie XS. (2012). "Genome-wide detection of single-nucleotide and copy-number variations of a single human cell". Science. 338 (6114): 1622–6. Bibcode:2012Sci...338.1622Z. doi:10.1126/science.1229164. PMC 3600412. PMID 23258894.
- ↑ Kurimoto, K.; Yabuta Y; Ohinata Y; Saitou M. (2007). "Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis". Nat Protoc. 2 (3): 739–52. doi:10.1038/nprot.2007.79. PMID 17406636.
- ↑ Yachida, S.; Jones S; Bozic I; et al. (2010). "Distant metastasis occurs late during the genetic evolution of pancreatic cancer". Nature. 467 (7319): 1114–7. Bibcode:2010Natur.467.1114Y. doi:10.1038/nature09515. PMC 3148940. PMID 20981102.
- ↑ Frumkin, D.; Wasserstrom A; Itzkovitz S; Harmelin A; Rechavi G; Shapiro E. (2008). "Amplification of multiple genomic loci from single cells isolated by laser micro-dissection of tissues". BMC Biotechnol. 8 (17). doi:10.1186/1472-6750-8-17. PMC 2266725. PMID 18284708.
- ↑ Dalerba, P.; Kalisky T; Sahoo D; et al. (2011). "Single-cell dissection of transcriptional heterogeneity in human colon tumors". Nat. Biotechnol. 29 (12): 1120–7. doi:10.1038/nbt.2038. PMC 3237928. PMID 22081019.
- ↑ White, AK.; VanInsberghe M; Petriv OI; et al. (2011). "High-throughput microfluidic single-cell RT-qPCR". Proc Natl Acad Sci U S A. 108 (34): 13999–4004. Bibcode:2011PNAS..10813999W. doi:10.1073/pnas.1019446108. PMC 3161570. PMID 21808033.