Proteogenomics
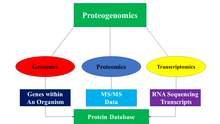
Proteogenomics is a field of biological research that utilizes a combination of proteomics, genomics, and transcriptomics to aid in the discovery and identification of peptides. Protegenomics is used to identify new peptides by comparing MS/MS spectra against a protein database that has been derived from genomic and trancriptomic information. Proteogenomics often refers to studies that use proteomic information, often derived from mass spectrometry, to improve gene annotations.[1] Genomics deals with the genetic code of entire organisms, while transcriptomics deals with the study of RNA sequencing and transcripts. Proteomics utilizes tandem mass spectrometry and liquid chromatography to identify and study the functions of proteins. Proteomics is being utilized to discover all the proteins expressed within an organism, known as its proteome.[2] The issue with proteomics is that it relies on the assumption that current gene models are correct and that the correct protein sequences can be found using a reference protein sequence database; however, this is not always the case as some peptides cannot be located in the database. In addition, novel protein sequences can occur through mutations. these issues can be fixed with the use of proteomic, genomic, and trancriptomic data. The utilization of both proteomics and genomics led to proteogeonmics which became its own field in 2004.[1][3][4]
Methodology

The main idea behind the proteogenomic approach is to identify peptides by comparing MS/MS data to protein databases that contain predicted protein sequences. The protein database is generated in a variety of ways through the utilization of genomic and transcriptomic data. Below are some of the ways in which protein databases are generated:
Six-frame translation
Six-frame translations can be utilized to generate a database that predicts protein sequences. The limitation of this method is that databases will be very large due to the number of sequences that are generated, some of which do not exist in nature. [1]
Ab initio gene prediction
In this method, a protein base is generated by gene predicting algorithms that enable the identification of protein coding regions. The database is similar to one generated through six-frame translation in regards to the fact that the databases can be very large.[1]
Expressed sequence tag data
Six-frame translations can utilize an expressed sequence tag (EST) to generate protein databases. EST data provide transcription information that can aid in the creation of the database. The database can be very large and has the disadvantage of having multiple copies of a given sequence present; however, this problem can be circumvented by compressing the protein sequence generated through computational strategies.[1]
Other methods
Protein databases can also be created by using RNA sequencing data, annotated RNA transcripts, and variant protein sequences. Also, there are other more specialized protein databases that can be made to appropriately identify the peptide of interest.[1]
Another method in the identification of proteins through proteogenomics is comparative proteogenomics. Comparitive proteogenomics compares proteomic data from multiple related species concurrently and exploits the homology between their proteins to improve annotations with higher statistical confidence.[5][6]
Applications
Proteogenomics can be applied in different ways. One application is the improvement of gene annotations in various organisms. Gene annotation involves discovering genes and their functions[7]. Proteogenomics has become especially useful in the discovery and improvement of gene annotations in prokaryotic organisms. For example, various microorganisms have had their genomic annotation studied through the proteogenomic approach including, Escherichia coli, Mycobacterium, and multiple species of Shewanella bacteria. [8]
Besides improving gene annotations, proteogenomic studies can also provide valuable information about the presence of programmed frameshifts, N-terminal methionine excision, signal peptides, proteolysis and other post-translational modifications.[3][5] Proteogenomics has potential applications in medicine, especially to oncology research. Cancer occurs through genetic mutations such as methylation, translocation, and somatic mutations. Research has shown that both genomic and proteomic information are needed to understand the molecular variations that lead to cancer.[2][9] Proteogenomics has aided in this through the identification of protein sequences that may have functional roles in cancer.[10] A specific example of this occurred in a study involving colon cancer that resulted in the discovery of potential targets for cancer treatment.[2] Proteogenomics has also led to personalized cancer targeting immunotherapies, where antibody epitopes for cancer antigens are predicted using proteogenomics to create medicines that act on the patient's specific tumor.[11] In addition to treatment, proteogenonomics may provide insight into cancer diagnosis. In studies involving colon and rectal cancer, proteogenomics was utilized to identify somatic mutations. The identification of somatic mutations in patients could be used to diagnose cancer in patients. In addition to direct applications in cancer treatment and diagnosis, a proteogenomic approach can be used to study proteins that result in resistance to chemotherapy. [9]
Challenges
Proteogenomics may offer methods of peptide identification without having the disadvantage of incomplete or inaccurate protein databases faced by proteomics;however, there are incurring challenges with the proteogenomic approach.[1] One of the biggest challenges of proteogenomics is the sheer size of protein databases generated. statistically, a large protein database is more likely to result in the incorrect matching of the data from the protein database to the MS/MS data, this issue can hinder the identification of new peptides. False positives are also an issue through proteogenomic approaches. false positives can occur as a result of extremely large protein data bases where miss-matched data leads to incorrect identification. Another issue is the incorrect matching of MS/MS spectra to protein sequence data that corresponds to a similar peptide instead of the actual peptide. There are cases of receiving data of a peptide located at multiple gene sites, this can lead to data that can be interpreted in different ways. Despite these challenges, there are ways to reduce many of the errors that occur. For example, when dealing with a very large protein database, one could compare the identified novel peptide sequences to all of the sequences within the database and then compare the post transnational modifications. Next it can be determined if the two sequences represent the same peptide or if they are two different peptides.[1]
References
- 1 2 3 4 5 6 7 8 Nesvizhskii, Alexey I (1 November 2014). "Proteogenomics: concepts, applications and computational strategies". Nature Methods. 11 (11): 1114–1125. doi:10.1038/nmeth.3144. PMC 4392723.
- 1 2 3 Sajjad, Wasim; Rafiq, Muhammad; Ali, Barkat; Hayat, Muhammad; Zada, Sahib; Sajjad, Wasim; Kumar, Tanweer (July 2016). "Proteogenomics: New Emerging Technology". HAYATI Journal of Biosciences. 23 (3): 97–100. doi:10.1016/j.hjb.2016.11.002.
- 1 2 Gupta N., Tanner S., Jaitly N., Adkins J.N., Lipton M., Edwards R., Romine M., Osterman A., Bafna V., Smith R.D., et al. Whole proteome analysis of post-translational modifications: Applications of mass-spectrometry for proteogenomic annotation. Genome Res. 2007;17:1362–1377.
- ↑ . Ansong C., Purvine S. O., Adkins J. N., Lipton M. S., Smith R. D. ( 2008) Proteogenomics: needs and roles to be filled by proteomics in genome annotation. Brief. Funct. Genomics Proteomics 7, 50– 62.
- 1 2 Gupta N., Benhamida J., Bhargava V., Goodman D., Kain E., Kerman I., Nguyen N., Ollikainen N., Rodriguez J., Wang J., et al. Comparative proteogenomics: Combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res. 2008;18:1133–1142.
- ↑ Gallien S., Perrodou E., Carapito C., Deshayes C., Reyrat J. M., Van Dorsselaer A., Poch O., Schaeffer C., Lecompte O. ( 2009) Ortho-proteogenomics: multiple proteomes investigation through orthology and a new MS-based protocol. Genome Res 19, 128– 135.
- ↑ Ansong, C.; Purvine, S. O.; Adkins, J. N.; Lipton, M. S.; Smith, R. D. (7 March 2008). "Proteogenomics: needs and roles to be filled by proteomics in genome annotation". Briefings in Functional Genomics and Proteomics. 7 (1): 50–62. doi:10.1093/bfgp/eln010. PMID 18334489.
- ↑ Kucharova, Veronika; Wiker, Harald G. (December 2014). "Proteogenomics in microbiology: Taking the right turn at the junction of genomics and proteomics". Proteomics. 14 (23–24): 2360–2675. doi:10.1002/pmic.201400168.
- 1 2 Shukla, Hem D.; Mahmood, Javed; Vujaskovic, Zeljko (December 2015). "Integrated proteo-genomic approach for early diagnosis and prognosis of cancer". Cancer Letters. 369 (1): 28–36. doi:10.1016/j.canlet.2015.08.003. PMID 26276717.
- ↑ Chambers, Matthew C.; Jagtap, Pratik D.; Johnson, James E.; McGowan, Thomas; Kumar, Praveen; Onsongo, Getiria; Guerrero, Candace R.; Barsnes, Harald; Vaudel, Marc (2017-11-01). "An Accessible Proteogenomics Informatics Resource for Cancer Researchers". Cancer Research. 77 (21): e43–e46. doi:10.1158/0008-5472.can-17-0331. PMID 29092937.
- ↑ Creech, Amanda L.; Ting, Ying S.; Goulding, Scott P.; Sauld, John FK; Barthelme, Dominik; Rooney, Michael S.; Addona, Terri A.; Abelin, Jennifer G. (2018). "The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction". Proteomics: n/a–n/a. doi:10.1002/pmic.201700259. ISSN 1615-9861.