Perfect phylogeny
Perfect phylogeny is a term used in computational phylogenetics to denote a phylogenetic tree in which all internal nodes may be labeled such that all characters evolve down the tree without homoplasy. That is, characteristics do not hold to evolutionary convergence, and do not have analogous structures. Statistically, this can be represented as an ancestor having state "0" in all characteristics where 0 represents a lack of that characteristic. Each of these characteristics changes from 0 to 1 exactly once and never reverts to state 0. It is rare that actual data adheres to the concept of perfect phylogeny.[1][2]
Building a Phylogenetic Tree
In general there are two different data types that are used in the construction of a phylogenetic tree. In distance-based computations a phylogenetic tree is created by analyzing relationships among the distance between species and the edge lengths of a corresponding tree. Using a character-based approach employs character states across species as an input in an attempt to find the most "perfect" phylogenetic tree.[3][4]
The statistical components of a perfect phylogenetic tree can best be described as follows:[3]
A perfect phylogeny for an n x m character state matrix M is a rooted tree T with n leaves satisfying:
i. Each row of M labels exactly one leaf of T
ii. Each column of M labels exactly one edge of T
iii. Every interior edge of T is labeled by at least one column of M
iv. The characters associated with the edges along the unique path from root to a leaf v exactly specify the character vector of v, i.e. the character vector has a 1 entry in all columns corresponding to characters associated to path edges and a 0 entry otherwise.
It is worth noting that it is very rare to find actual phylogenetic data that adheres to the concepts and limitations detailed here. Therefore, it is often the case that researchers are forced to compromise by developing trees that simply try to minimize homoplasy, finding a maximum-cardinality set of compatible characters, or constructing phylogenies that match as closely as possible to the partitions implied by the characters.
Example
Both of these data sets illustrate examples of character state matrices. Using matrix M'1 one is able to observe that the resulting phylogenetic tree can be created such that each of the characters label exactly one edge of the tree. In contrast, when observing matrix M'2, one can see that there is no way to set up the phylogenetic tree such that each character labels only one edge length.[3]
- Character state matrices
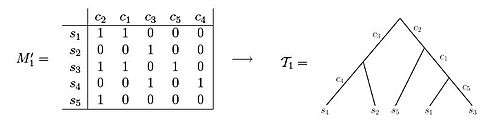 An example of a character matrix that can be depicted as a perfect phylogeny
An example of a character matrix that can be depicted as a perfect phylogeny
Usage
Perfect phylogeny is a theoretical framework that can also be used in more practical methods. One such example is that of Incomplete Directed Perfect Phylogeny. This concept involves utilizing perfect phylogenies with real, and therefore incomplete and imperfect, datasets. Such a method utilizes SINEs to determine evolutionary similarity. These Short Interspersed Elements are present across many genomes and can be identified by their flanking sequences. SINEs provide information on the inheritance of certain traits across different species. Unfortunately, if a SINE is missing it is difficult to know whether those SINEs were present prior to the deletion. By utilizing algorithms derived from perfect phylogeny data we are able to attempt to reconstruct a phylogenetic tree in spite of these limitations.[5]
Perfect phylogeny is also used in the construction of haplotype maps. By utilizing the concepts and algorithms described in perfect phylogeny one can determine information regarding missing and unavailable haplotype data.[6] By assuming that the set of haplotypes that result from genotype mapping corresponds and adheres to the concept of perfect phylogeny (as well as other assumptions such as perfect Mendelian inheritance and the fact that there is only one mutation per SNP), one is able to infer missing haplotype data.[7][8][9] [10]
External links
- One of several programs available for analysis and creation of phylogenetic trees
- Another such program for phylogenetic tree analysis
- Additional program for tree analysis
- A paper detailing an example of how perfect phylogeny can be utilized outside of the field of genetics, as in language association
References
- ↑ Fernandez-Baca, David. "The Perfect Phylogeny Problem" (PDF). Kluwer Academic Publishers. Retrieved 30 September 2012.
- ↑ Nakhleh, Luay; et al. "Perfect Phylogenetic Networks: A New Methodology for Reconstructing the Evolutionary History of Natural Languages" (PDF). Retrieved 1 October 2012.
- 1 2 3 Uhler, Caroline. "Finding a Perfect Phylogeny" (PDF). Retrieved 29 September 2012.
- ↑ Nikaido, Masato; et al. "Phylogenetic relationships among cetartiodactyls based on insertions of short and long interpersed elements: Hippopotamuses are the closest extant relatives of whales". Retrieved 30 September 2012.
- ↑ Pe'er, I.; et al. "Incomplete Directed Perfect Phylogeny". Tel-Aviv University. Archived from the original on 20 October 2013. Retrieved 30 October 2012.
- ↑ Eskin, Eleazar; et al. "Efficient Reconstruction of Haplotype Structure via Perfect Phylogeny" (PDF). University of California, Berkeley. Retrieved 30 October 2012.
- ↑ Gusfield, Dan. "An Overview of Computational Methods for Haplotype Inference" (PDF). University of California, Davis. Retrieved 18 November 2012.
- ↑ Ding, Zhihong; et al. "A Linear Time Algorithm for the Perfect Phylogeny Haplotyping Problem". University of California, Davis. Retrieved 18 November 2012.
- ↑ Bafna, V.; et al. "Haplotyping as Perfect Phylogeny: a direct approach". J Comput Biol. 10: 323–40. doi:10.1089/10665270360688048. PMID 12935331.
- ↑ Seyalioglu, Hakan. "Haplotyping as Perfect Phylogeny" (PDF). Retrieved 30 October 2012.