Morquio syndrome
| Morquio Syndrome | |
|---|---|
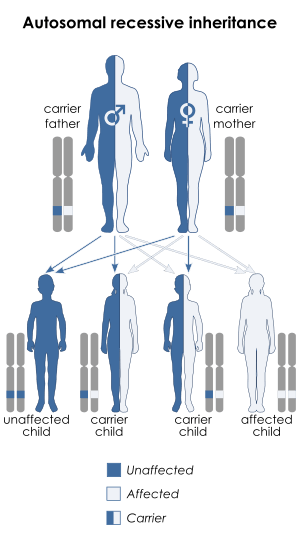 | |
| Morquio syndrome is inherited via an autosomal recessive manner | |
| Specialty |
Endocrinology |
Morquio syndrome (referred to as mucopolysaccharidosis IV, MPS IV, Morquio-Brailsford syndrome, or Morquio)[1] is a rare metabolic disorder in which the body cannot process certain types of mucopolysaccharides (long chains of sugar molecules), which the body uses as lubricants and shock absorbers. This birth defect, which is autosomal recessive, is thus a lysosomal storage disorder that is usually inherited.[2]:544 In the US, the incidence rate for Morquio is estimated at between 1 in 200,000 and 1 in 300,000 live births.[1]
The build-up or elimination of mucopolysaccharides, rather than processing by their usual biochemical pathways, causes various symptoms. These involve accumulation of keratan sulfate.[3]
Signs and symptoms
The following signs are associated with the disease
- Abnormal heart development
- Abnormal skeletal development
- Hypermobile joints
- Large fingers
- Knock-knees
- Widely spaced teeth
- Bell-shaped chest (flared ribs)
- Compression of spinal cord
- Enlarged heart
- Dwarfism
- Heart murmur
- below average height for certain age
Patients with Morquio syndrome appear healthy at birth.[1] They often present with spinal deformity, and there is growth retardation and possibly genu valgum in the second or third year of life. A patient with Morquio's syndrome is likely to die at an early age. Symptoms of the disease may include:
- Short stature and short neck (caused by flat vertebrae)
- Moderate kyphosis or scoliosis
- Mild pectus carinatum ("pigeon chest")
- Cervical spine: odontoid hypoplasia, atlanto-axial instability; may be associated with myelopathy with gradual loss of walking ability
- Joint laxity, mild dysostosis multiplex, dysplastic hips, large unstable knees, large elbows and wrists, and flat feet
- The combined abnormalities usually result in a duck-waddling gait
- Mid-face hypoplasia and mandibular protrusion
- Thin tooth enamel
- Corneal clouding
- Mild hepatosplenomegaly
Regarding the life span of people with Morquio, some can die as early as 2 or 3 years old, and some can live up to 60 or 70 years old. The oldest known person with Morquio syndrome type IV A was Kenneth D. Martin, who was born in Osage City, Kansas, USA and was 81 years old at the time of his death [4]
Diagnosis
Classification
This syndrome has two forms, A and B, referred to as Morquio A and Morquio B syndrome or MPA IVA and MPS IVB.[5] The two forms are distinguished by the gene product involved; A involves a malfunction in the GALNS gene product (galactosamine-6 sulfatase), while B involves a malfunction of the GLB1 gene product (beta-galactosidase).
| MPS-III type | gene | enzyme | chromosomal region |
|---|---|---|---|
| MPS-IV A | GALNS | galactosamine-6-sulfate sulfatase | 16q24 |
| MPS-IV B | GLB1 | Beta-galactosidase | 3p22 |
The human GLB1 gene, located at the chromosomal band 3p22.3, encodes beta-galactosidase-1, a lysosomal hydrolase that cleaves the terminal beta-galactose from ganglioside substrates and other glycoconjugates. Loss of function alleles typically cause a spectrum of the condition GM1-gangliosidosis. However, specific alleles, including combinations with most commonly the W273L variant can cause Morquio B syndrome.
Treatment
The treatment for Morquio syndrome consists of prenatal identification and of enzyme replacement therapy. On 12 February 2014, the US Food and Drug Administration approved the drug elosulfase alfa (Vimizim) for treating the disease.[6]
Life expectancy
Mean age at death (±standard deviation) was 25.30 ± 17.43 years, with female patients living longer than male patients (26.55 ± 12.28 years versus 22.95 ± 17.63 years, respectively). Respiratory failure was the primary cause of death in nearly two-thirds of patients (63%). Other causes of death were cardiac failure (11%), post-traumatic organ failure (11%), complications of surgery (11%) and myocardial infarction (4%). Life expectancy increased gradually over time (R2 = 0.0963), and mean age at death due to respiratory failure improved from 17.42 ± 9.54 years in the 1980s to 30.74 ± 10.84 years in the 2000
History
The condition was first described, simultaneously and independently, in 1929, by Luis Morquio (1867–1935),[7] a prominent Uruguayan physician who discovered it in Montevideo, and James Frederick Brailsford (1888–1961), an English radiographer in Birmingham, England.[8][9] They both recognized the occurrence of corneal clouding, aortic valve disease, and urinary excretion of keratan sulfate. Morquio observed the disorder in four siblings in a family of Swedish extraction and reported his observations in French.
See also
References
- 1 2 3 "MPS IV (Morquio syndrome)". MPSSociety.org. National MPS Society. Retrieved 14 January 2015.
- ↑ James, William D.; Berger, Timothy G. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ Prat C, Lemaire O, Bret J, Zabraniecki L, Fournié B (May 2008). "Morquio syndrome: Diagnosis in an adult". Joint Bone Spine. 75 (4): 495–8. doi:10.1016/j.jbspin.2007.07.021. PMID 18456538.
- ↑ "http://och-c.com/?q=content/kenneth-dean-martin"
- ↑ Classification information from OMIM records 253000 and 253010.
- ↑ "FDA approves Vimizim to treat rare congenital enzyme disorder" (Press release). US Food and Drug Administration. 14 February 2014. Retrieved 14 January 2015.
- ↑ Morquio, L. (1929). "Sur une forme de dystrophie osseuse familiale". Archives de médecine des infants. Paris. 32: 129–135. ISSN 0365-4311.
- ↑ synd/2108 at Who Named It?
- ↑ Brailsford, J. F. (1929). "Chondro-osteo-dystrophy: Roentgenographic & clinical features of a child with dislocation of vertebrae". American Journal of Surgery. New York. 7 (3): 404–410. doi:10.1016/S0002-9610(29)90496-7.
External links
| Classification | |
|---|---|
| External resources |