Maroteaux–Lamy syndrome
| Maroteaux–Lamy syndrome | |
|---|---|
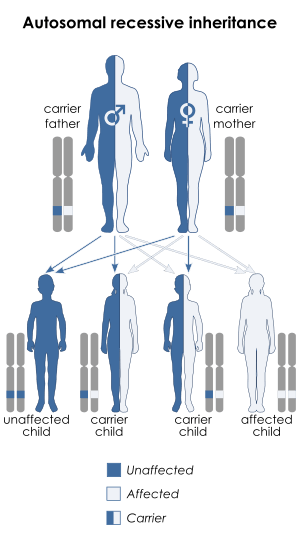 | |
| This condition is inherited in an autosomal recessive manner | |
| Specialty |
Endocrinology |
Maroteaux–Lamy syndrome (also known as mucopolysaccharidosis type VI,[1] MPS VI, or polydystrophic dwarfism) is a form of mucopolysaccharidosis caused by a deficiency in arylsulfatase B (ARSB).[2] It is named after Pierre Maroteaux (1926–) and his mentor Maurice Emil Joseph Lamy (1895–1975), both French physicians.[3][4]
Symptoms
Children with Maroteaux–Lamy syndrome usually have normal intellectual development but share many of the physical symptoms found in Hurler syndrome. Caused by the deficient enzyme N-acetylgalactosamine 4-sulfatase, Maroteaux–Lamy syndrome has a variable spectrum of severe symptoms. Neurological complications include clouded corneas, deafness, thickening of the dura (the membrane that surrounds and protects the brain and spinal cord), and pain caused by compressed or traumatized nerves and nerve roots.
Signs are revealed early in the affected child's life, with one of the first symptoms often being a significantly prolonged age of learning how to walk.[5] By age 10 children have developed a shortened trunk, crouched stance, and restricted joint movement. In more severe cases, children also develop a protruding abdomen and forward-curving spine. Skeletal changes (particularly in the pelvic region) are progressive and limit movement. Many children also have umbilical hernia or inguinal hernias. Nearly all children have some form of heart disease, usually involving valve dysfunction. An enzyme replacement therapy, galsulfase (Naglazyme), was tested on patients with Maroteaux–Lamy syndrome and was successful in that it improved growth and joint movement. An experiment was then carried out to see whether an injection of the missing enzyme into the hips would help the range of motion and pain. At a cost of $365,000 a year, Naglazyme is one of the world's most expensive drugs.[6]
Treatment
Enzyme replacement therapy: Naglazyme
See also
References
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- ↑ Garrido E, Cormand B, Hopwood JJ, Chabás A, Grinberg D, Vilageliu L (July 2008). "Maroteaux-Lamy syndrome: functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene". Mol. Genet. Metab. 94 (3): 305–12. doi:10.1016/j.ymgme.2008.02.012. PMID 18406185.
- ↑ synd/1619 at Who Named It?
- ↑ MAROTEAUX P, LEVEQUE B, MARIE J, LAMY M (September 1963). "[A NEW DYSOSTOSIS WITH URINARY ELIMINATION OF CHONDROITIN SULFATE B.]". Presse Med (in French). 71: 1849–52. PMID 14091597.
- ↑ "Topic Galleries". Chicago Tribune.
- ↑ Larry Schwartz. "The 9 Most Expensive Medicines in the World–Courtesy of Big Pharma". AlterNet.
External links
| Classification | |
|---|---|
| External resources |