Joubert syndrome
| Joubert syndrome | |
|---|---|
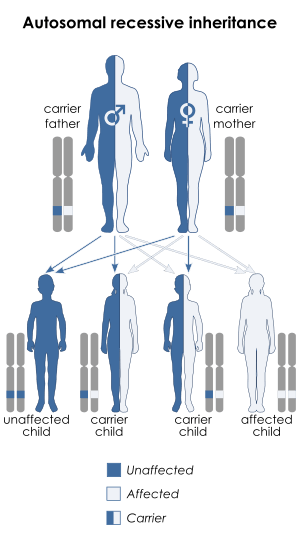 | |
| Joubert syndrome is inherited via an autosomal recessive manner | |
| Specialty |
Medical genetics |
Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.
Joubert syndrome is one of the many genetic syndromes associated with syndromic retinitis pigmentosa.[1] The syndrome was first identified in 1969 by pediatric neurologist Marie Joubert in Montreal, Quebec, Canada, while working at the Montreal Neurological Institute and McGill University.[2]
Signs and symptoms
Most of the signs and symptoms of the Joubert syndrome appear very early in infancy with most children showing delays in gross motor milestones.[3] Although other signs and symptoms vary widely from individual to individual, they generally fall under the hallmark of cerebellum involvement or in this case, lack thereof. Consequently, the most common features include ataxia (lack of muscle control), hyperpnea (abnormal breathing patterns), sleep apnea, abnormal eye and tongue movements, and hypotonia in early childhood. Other malformations such as polydactyly (extra fingers and toes), cleft lip or palate, tongue abnormalities, and seizures may also occur. Developmental delays, including cognitive, are always present to some degree.[4]
Those suffering from this syndrome often exhibit specific facial features such as a broad forehead, arched eyebrows, ptosis (droopy eyelids), hypertelorism (widely spaced eyes), low-set ears and a triangle shaped mouth. Additionally, this disease can include a broad range of other abnormalities to other organ systems such as retinal dystrophy, kidney diseases, liver diseases, skeletal deformities and endocrine (hormonal) problems.[5]
Genetics
A number of mutations have been identified in individuals with Joubert syndrome (JBTS) which allowed for classification of the disorder into subtypes.
This disorder can be caused by mutations in more than 30 genes within genetic makeup. The primary cilia play an important role in the structure and function of cells. When primary cilia are mutated and defected, it can cause various genetic disorders among individuals. This mutation of primary cilia can disrupt significant signaling pathways during the development of the fetus.
Mutations in these various genes are known for causing around 60-90% of Joubert Syndrome cases. The remaining cases, the cause is unknown if isn't linked to a mutation of known genes.[6]
| Type | OMIM | Gene | Locus | Inheritance | Remarks |
|---|---|---|---|---|---|
| JBTS1 | 213300 | INPP5E | 9q34.3 | Autosomal recessive | Also known as Cerebellooculorenal syndrome 1 (CORS1) |
| JBTS2 | 608091 | TMEM216 | 11q12.2 | Autosomal recessive | Also known as Cerebellooculorenal syndrome 2 (CORS2) |
| JBTS3 | 608629 | AHI1 | 6q23.3 | Autosomal recessive | |
| JBTS4 | 609583 | NPHP1 | 2q13 | ||
| JBTS5 | 610188 | CEP290 NPHP6 |
12q21.32 | Autosomal recessive | |
| JBTS6 | 610688 | TMEM67 | 8q22.1 | Autosomal recessive | |
| JBTS7 | 611560 | RPGRIP1L | 16q12.2 | ||
| JBTS8 | 612291 | ARL13B | 3q11.1 | ||
| JBTS9 | 612285 | CC2D2A | 4p15.32 | Autosomal recessive | |
| JBTS10 | 300804 | OFD1 | Xp22.2 | X-linked recessive | |
| JBTS11 | – | TTC21B | 2q24.3 | ||
| JBTS12 | – | KIF7 | 15q26.1 | ||
| JBTS13 | 614173 | TCTN1 | 12q24.11 | ||
| JBTS14 | 614424 | TMEM237 | 2q33.1 | Autosomal recessive | |
| JBTS15 | 614464 | CEP41 | 7q32.2 | Autosomal recessive | |
| JBTS16 | 614465 | TMEM138 | 11q12.2 | Autosomal recessive | |
| JBTS17 | 614615 | C5ORF42 | 5p13.2 | ||
| JBTS18 | 614815 | TCTN3 | 10q24.1 | ||
| JBTS19 | – | ZNF423 | 16q12.1 | Autosomal dominant | |
| JBTS20 | 614970 | TMEM231 | 16q23.1 | Autosomal recessive | |
| 611654 | CSPP1,[7][8][9] | 8q13.2 | Autosomal recessive | ||
| - | ARMC9 | 2q37.1 | Autosomal recessive |
Diagnosis
The disorder is characterized by absence or underdevelopment of the cerebellar vermis and a malformed brain stem (molar tooth sign), both of which can be visualized on a MRI scan.[10] Together with this sign, the diagnosis is based on the physical symptoms and genetic testing for mutations. If the gene mutations have been identified in a family member, prenatal or carrier diagnosis can be pursued.[3]
Joubert Syndrome is known to affect 1 in 80,000-100,000 newborns. Due to the variety of genes this disorder is affected by, it is likely to be under-diagnosed. It is commonly found in Ashkenazi Jewish, French-Canadians, and Hutterite ethnic populations. Most cases of Joubert syndrome are autosomal recessive - in these cases, both parents are either carriers or affected. Rarely, Joubert syndrome is inherited in an X-linked recessive pattern. In these cases, males are more commonly affected because affected males must have one X chromosome mutated, while affected females must have mutated genes on both X chromosomes.[6]
Treatment
Treatment for Joubert syndrome is symptomatic and supportive.[4] Infant stimulation and physical, occupational, speech and hearing therapy may benefit some patients. Infants with abnormal breathing patterns should be monitored.
The syndrome is associated with progressive worsening for kidneys, the liver and the eyes and thus require regular monitoring.[4]
Prognosis
In a sample of 19 children, a 1997 study found that 3 died before the age of 3, and 2 never learned to walk. The children had various levels of delayed development with developmental quotients from 60 to 85.[11]
Research
Research has revealed that a number of genetic disorders, not previously thought to be related, may indeed be related as to their root cause. Joubert syndrome is one such disease. It is a member of an emerging class of diseases called ciliopathies.
The underlying cause of the ciliopathies may be a dysfunctional molecular mechanism in the primary cilia structures of the cell, organelles which are present in many cellular types throughout the human body. The cilia defects adversely affect "numerous critical developmental signaling pathways" essential to cellular development and thus offer a plausible hypothesis for the often multi-symptom nature of a large set of syndromes and diseases.
Currently recognized ciliopathies include Joubert syndrome, primary ciliary dyskinesia (also known as Kartagener Syndrome), Bardet-Biedl syndrome, polycystic kidney disease and polycystic liver disease, nephronophthisis, Alström syndrome, Meckel-Gruber syndrome and some forms of retinal degeneration.[12]
Joubert syndrome type 2 is disproportionately frequent among people of Jewish descent.[13]
References
- ↑ Saraiva, JM; Baraitser, M (1992). "Joubert syndrome: a review". American Journal of Medical Genetics. 43 (4): 726–731. doi:10.1002/ajmg.1320430415. PMID 1341417.
- ↑ Joubert M, Eisenring JJ, Robb JP, Andermann F (September 1969). "Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation". Neurology. 19 (9): 813–25. doi:10.1212/wnl.19.9.813. PMID 5816874.
- 1 2 "Joubert Syndrome - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Retrieved 2016-12-19.
- 1 2 3 Parisi, Melissa A.; Doherty, Dan; Chance, Phillip F.; Glass, Ian A. (2007-03-21). "Joubert syndrome (and related disorders) (OMIM 213300)". European Journal of Human Genetics. 15 (5): 511–521. doi:10.1038/sj.ejhg.5201648. ISSN 1018-4813.
- ↑ Reference, Genetics Home. "Joubert syndrome". Genetics Home Reference. Retrieved 2016-12-19.
- 1 2 Reference, Genetics Home. "Joubert syndrome". Genetics Home Reference. Retrieved 2017-09-13.
- ↑ Shaheen, R.; Shamseldin, H. E.; Loucks, C. M.; Seidahmed, M. Z.; Ansari, S.; Ibrahim Khalil, M.; Al-Yacoub, N.; Davis, E. E.; Mola, N. A.; Szymanska, K.; Herridge, W.; Chudley, A. E.; Chodirker, B. N.; Schwartzentruber, J.; Majewski, J.; Katsanis, N.; Poizat, C.; Johnson, C. A.; Parboosingh, J.; Boycott, K. M.; Innes, A. M.; Alkuraya, F. S. (2013). "Mutations in CSPP1, Encoding a Core Centrosomal Protein, Cause a Range of Ciliopathy Phenotypes in Humans". The American Journal of Human Genetics. 94 (1): 73–9. doi:10.1016/j.ajhg.2013.11.010. PMC 3882732. PMID 24360803.
- ↑ Akizu, N.; Silhavy, J. L.; Rosti, R. O.; Scott, E.; Fenstermaker, A. G.; Schroth, J.; Zaki, M. S.; Sanchez, H.; Gupta, N.; Kabra, M.; Kara, M.; Ben-Omran, T.; Rosti, B.; Guemez-Gamboa, A.; Spencer, E.; Pan, R.; Cai, N.; Abdellateef, M.; Gabriel, S.; Halbritter, J.; Hildebrandt, F.; Van Bokhoven, H.; Gunel, M.; Gleeson, J. G. (2013). "Mutations in CSPP1 Lead to Classical Joubert Syndrome". The American Journal of Human Genetics. 94 (1): 80–6. doi:10.1016/j.ajhg.2013.11.015. PMC 3882909. PMID 24360807.
- ↑ Tuz, K.; Bachmann-Gagescu, R.; o’Day, D. R.; Hua, K.; Isabella, C. R.; Phelps, I. G.; Stolarski, A. E.; o’Roak, B. J.; Dempsey, J. C.; Lourenco, C.; Alswaid, A.; Bönnemann, C. G.; Medne, L.; Nampoothiri, S.; Stark, Z.; Leventer, R. J.; Topçu, M.; Cansu, A.; Jagadeesh, S.; Done, S.; Ishak, G. E.; Glass, I. A.; Shendure, J.; Neuhauss, S. C. F.; Haldeman-Englert, C. R.; Doherty, D.; Ferland, R. J. (2013). "Mutations in CSPP1 Cause Primary Cilia Abnormalities and Joubert Syndrome with or without Jeune Asphyxiating Thoracic Dystrophy". The American Journal of Human Genetics. 94 (1): 62–72. doi:10.1016/j.ajhg.2013.11.019. PMC 3882733. PMID 24360808.
- ↑ Brancati F, Dallapiccola B, Valente EM (2010). "Joubert Syndrome and related disorders". Orphanet J Rare Dis. 5: 20. doi:10.1186/1750-1172-5-20. PMC 2913941. PMID 20615230.
- ↑ Steinlin, M.; Schmid, M.; Landau, K.; Boltshauser, E. (1997-08-01). "Follow-Up in Children with Joubert Syndrome". Neuropediatrics. 28 (04): 204–211. doi:10.1055/s-2007-973701. ISSN 0174-304X. PMID 9309710.
- ↑ Badano, Jose L.; Norimasa Mitsuma; Phil L. Beales; Nicholas Katsanis (September 2006). "The Ciliopathies : An Emerging Class of Human Genetic Disorders". Annual Review of Genomics and Human Genetics. 7: 125–148. doi:10.1146/annurev.genom.7.080505.115610. PMID 16722803. Retrieved 2008-06-15.
- ↑ Gutkind, Lee; Kennedy, Pagan (10 October 2013). An Immense New Power to Heal: The Promise of Personalized Medicine. Underland Press. p. 36. ISBN 978-1-937163-07-5.
External links
| Classification | |
|---|---|
| External resources |