Bioconjugation
Bioconjugation is a chemical strategy to form a stable covalent link between two molecules, at least one of which is a biomolecule.
Function
Recent advances in the understanding of biomolecules enabled their application to numerous fields like medicine and materials. Synthetically modified biomolecules can have diverse functionalities, such as tracking cellular events, revealing enzyme function, determining protein biodistribution, imaging specific biomarkers, and delivering drugs to targeted cells.[1][2][3][4] Bioconjugation is a crucial strategy that links these modified biomolecules with different substrates.
Synthesis
Synthesis of bioconjugates involves a variety of challenges, ranging from the simple and nonspecific use of a fluorescent dye marker to the complex design of antibody drug conjugates.[1][3] As a result, various bioconjugation reactions – chemical reactions connecting two biomolecules together – have been developed to chemically modify proteins. Common types of bioconjugation reactions are coupling of lysine amino acid residues, coupling of cysteine residues, coupling of tyrosine residues, modification of tryptophan residues, and modification of the N- and C- terminus.[1][3][4]
However, these reactions often lack chemoselectivity and efficiency, because they depend on the presence of native amino acid residues, which are usually present in large quantities that hinder selectivity. There is an increasing need for chemical strategies that can effectively attach synthetic molecules site specifically to proteins. One strategy is to first install a unique functional group onto a protein, and then a bioorthogonal or click type reaction is used to couple a biomolecule with this unique functional group.[1] The bioorthogonal reactions targeting non-native functional groups are widely used in bioconjugation chemistry. Some important reactions are modification of ketone and aldehydes, Staudinger ligation with azides, copper-catalyzed Huisgen cycloaddition of azides, and strain promoted Huisgen cycloaddition of azides.[5][6][7][8]
Common bioconjugation reactions
The most common bioconjugations are coupling of a small molecule (such as biotin or a fluorescent dye) to a protein, or protein-protein conjugations, such as the coupling of an antibody to an enzyme.[9] Other less common molecules used in bioconjugation are oligosaccharides, nucleic acids, synthetic polymers such as polyethylene glycol,[10] and carbon nanotubes.[11] Antibody-drug conjugates such as Brentuximab vedotin and Gemtuzumab ozogamicin are also examples of bioconjugation, and are an active area of research in the pharmaceutical industry.[12] Recently, bioconjugation has also gained importance in nanotechnology applications such as bioconjugated quantum dots.
Reactions of lysine residues
The nucleophilic lysine residue is commonly targeted site in protein bioconjugation, typically through amine-reactive N-Hydroxysuccinimidyl (NHS) esters.[3] To obtain optimal number of deprotonated lysine residues, the pH of the aqueous solution must be below the pKa of the lysine ammonium group, which is around 10.5, so the typical pH of the reaction is about 8 and 9. The common reagent for the coupling reaction is NHS-ester (shown in the first reaction below in Figure 1), which reacts with nucleophilic lysine through a lysine acylation mechanism. Other similar reagents are isocyanates and isothiocyanates that undergo a similar mechanism (shown in the second and third reactions in Figure 1 below).[1] Benzoyl fluorides (shown in the last reaction below in Figure 1), which allows for lysine modification of proteins under mild conditions (low temperature, physiological pH), were recently proposed as an alternative to classically used lysine specific reagents.[13]
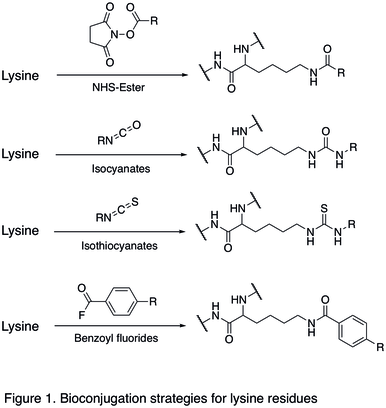
Reactions of cysteine residues
Because free cysteine rarely occurs on protein surface, it is an excellent choice for chemoselective modification.[14] Under basic condition, the cysteine residues will be deprotonated to generate a thiolate nucleophile, which will react with soft electrophiles, such as maleimides and iodoacetamides (shown in the first two reactions in Figure 2 below). As a result, a carbon-sulfur bond is formed. Another modification of cysteine residues involves the formation of disulfide bond (shown in the third reaction in Figure 2). The reduced cysteine residues react with exogenous disulfides, generating new disulfides bond on protein. An excess of disulfides is often used to drive the reaction, such as 2-thiopyridone and 3-carboxy-4-nitrothiophenol.[1][3] Electron-deficient alkynes were demonstrated to selectively react with cysteine residues of proteins in the presence of other nucleophilic amino acid residues. Depending on the alkyne substitution, these reactions can produce either cleavable (when alkynone derivatives are used),[15] or hydrolytically stable bioconjugates (when 3-arylpropiolonitriles are used; the last reaction below in Figure 2).[16]
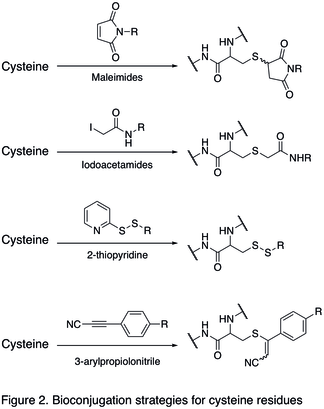
Reactions of tyrosine residues
Tyrosine residues are relatively unreactive; therefore they have not been a popular targets for bioconjugation. Recent development has shown that the tyrosine can be modified through electrophilic aromatic substitutions (EAS) reactions, and it is selective for the aromatic carbon adjacent to the phenolic hydroxyl group.[1] This becomes particularly useful in the case that cysteine residues cannot be targeted. Specifically, diazonium effectively couples with tyrosine residues (diazonium salt shown as reagent in the first reaction in Figure 3 below), and an electron withdrawing substituent in the 4-position of diazonium salt can effectively increase the efficiency of the reaction. Cyclic diazodicarboxyamide derivative like 4-Phenyl-1,2,4-triazole-3,5-dione (PTAD) were reported for selective bioconjugation on tyrosine residues (the second reaction in Figure 3 below).[17] A three-component Mannich-type reaction with aldehydes and anilines (the last reaction in Figure 3) was also described to be relatively tyrosine-selective under mild optimised reaction conditions.[18]
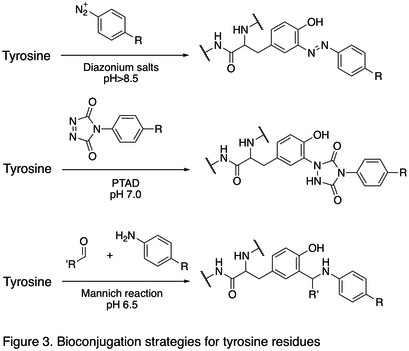
Reactions of N- and C- termini
Since natural amino acid residues are usually present in large quantities, it is often difficult to modify one single site. Strategies targeting the termini of protein have been developed, because they greatly enhanced the site selectivity of protein modification. One of the N- termini modifications involves the functionalization of the terminal amino acid. The oxidation of N-terminal serine and threonine residues are able to generate N-terminal aldehyde, which can undergo further bioorthogonal reactions (shown in the first reaction in Figure 4). Another type of modification involves the condensation of N-terminal cysteine with aldehyde, generating thiazolidine that is stable at high pH (second reaction in Figure 4). Using pyridoxal phosphate (PLP), several N-terminal amino acids can undergo transamination to yield N-terminal aldehyde, such as glycine and aspartic acid (third reaction in Figure 4).
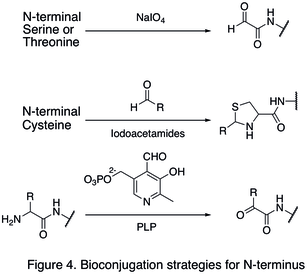
An example of C-termini modification is the native chemical ligation (NCL), which is the coupling between a C-terminal thioester and a N-terminal cysteine (Figure 5).
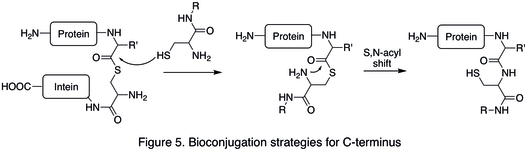
Bioorthogonal reactions
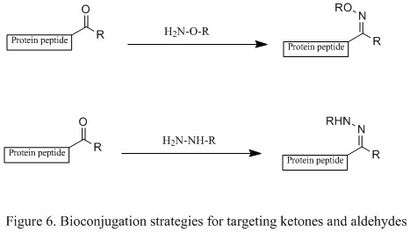
Modification of ketones and aldehydes
A ketone or aldehyde can be attached to a protein through the oxidation of N-terminal serine residues or transamination with PLP. Additionally, they can be introduced by incorporating unnatural amino acids via the Tirrell method or Schultz method.[5] They will then selectively condense with an alkoxyamine and a hydrazine, producing oxime and hydrazone derivatives (shown in the first and second reactions, respectively, in Figure 6). This reaction is highly chemoselective in terms of protein bioconjugation, but the reaction rate is slow. The mechanistic studies show that the rate determining step is the dehydration of tetrahedral intermediate, so a mild acidic solution is often employed to accelerate the dehydration step.[2]
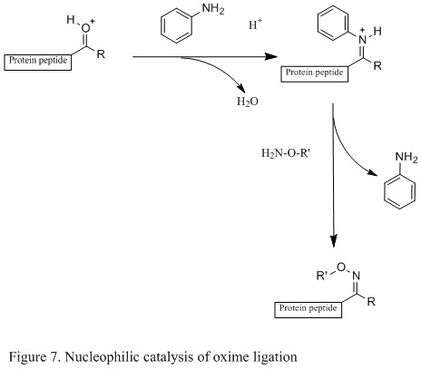
The introduction of nucleophilic catalyst can significantly enhance reaction rate (shown in Figure 7). For example, using aniline as a nucleophilic catalyst, a less populated protonated carbonyl becomes a highly populated protonated Schiff base.[19] In other words, it generates a high concentration of reactive electrophile. The oxime ligation can then occur readily, and it has been reported that the rate increased up to 400 times under mild acidic condition.[19] The key of this catalyst is that it can generate a reactive electrophile without competing with desired product.
Recent developments that exploit proximal functional groups have enabled hydrazone condensations[20] to operate at 20 M−1s−1 at neutral pH while oxime condensations have been discovered which proceed at 500-10000 M−1s−1 at neutral pH without added catalysts.[21][22]
Staudinger ligation with azides
The Staudinger ligation of azides and phosphine has been used extensively in field of chemical biology. Because it is able to form a stable amide bond in living cells and animals, it has been applied to modification of cell membrane, in vivoimaging, and other bioconjugation studies.[23][24][25][26]
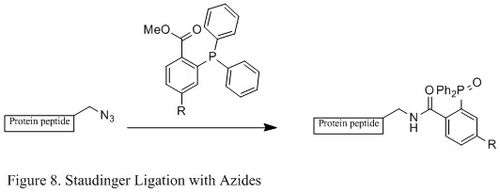
Contrasting with the classic Staudinger reaction, Staudinger ligation is a second order reaction in which the rate-limiting step is the formation of phosphazide (specific reaction mechanism shown in Figure 9). The triphenylphosphine first reacts with the azide to yield an azaylide through a four-membered ring transition state, and then an intramolecular reaction leads to the iminophosphorane intermediate, which will then give the amide-linkage under hydrolysis.[27]
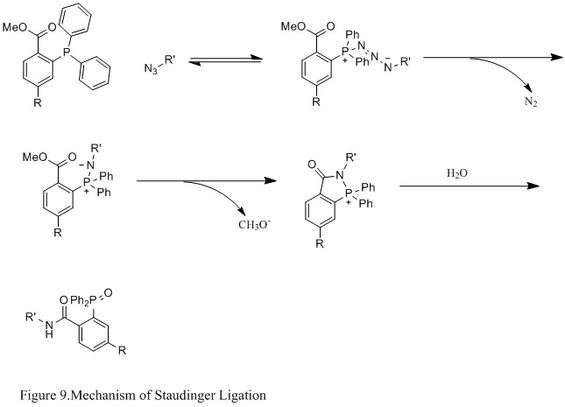
Huisgen cyclization of azides
Copper catalyzed Huisgen cyclization of azides
Azide has become a popular target for chemoselective protein modification, because they are small in size and has a favorable thermodynamic reaction potential. One such azide reactions is the [3+2] cycloaddition reaction with alkyne, but the reaction requires high temperature and often gives mixtures of regioisomers.
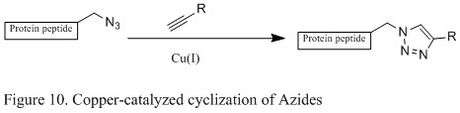
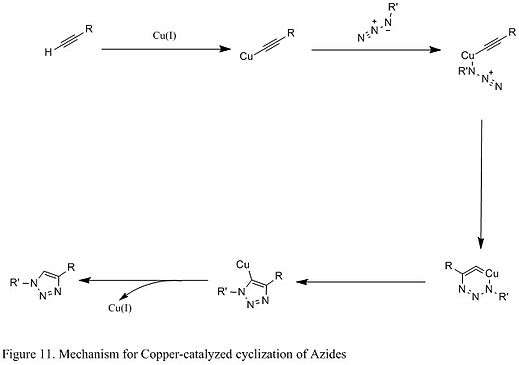
An improved reaction developed by chemist Karl Barry Sharpless involves the copper (I) catalyst, which couples azide with terminal alkyne that only give 1,4 substituted 1,2,3 triazoles in high yields (shown below in Figure 11). The mechanistic study suggests a stepwise reaction.[8] The Cu (I) first couples with acetylenes, and then it reacts with azide to generate a six-membered intermediate. The process is very robust that it occurs at pH ranging from 4 to 12, and copper (II) sulfate is often used as a catalyst in the presence of a reducing agent.[8]
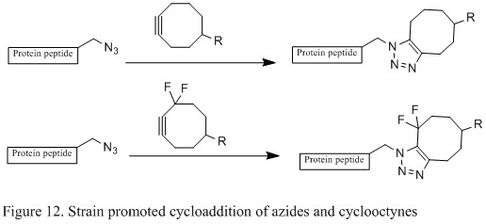
Strain promoted Huisgen cyclization of azides
Even though Staudinger ligation is a suitable bioconjugation in living cells without major toxicity, the phosphine’s sensitivity to air oxidation and its poor solubility in water significantly hinder its efficiency. The copper (I) catalyzed azide-alkyne coupling has reasonable reaction rate and efficiency under physiological conditions, but copper poses significant toxicity and sometimes interferes with protein functions in living cells. In 2004, chemist Carolyn R. Bertozzi’s lab developed a metal free [3+2] cycloaddition using strained cyclooctyne and azide. Cyclooctyne, which is the smallest stable cycloalkyne, can couple with azide through [3+2] cycloaddition, leading to two regioisomeric triazoles (Figure 12).[6] The reaction occurs readily at room temperature and therefore can be used to effectively modify living cells without negative effects. It has also been reported that the installation of fluorine substituents on a cyclic alkyne can greatly accelerate the reaction rate.[2][28]
See also
References
- 1 2 3 4 5 6 7 Stephanopoulos, N.; Francis, M. B. (2011). "Choosing an effective protein bioconjugation strategy". Nature Chemical Biology. 7 (12): 876–884. doi:10.1038/nchembio.720. PMID 22086289.
- 1 2 3 Tilley, S. D.; Joshi, N. S.; Francis, M. B. (2008). "Proteins: Chemistry and Chemical Reactivity". Wiley Encyclopedia of Chemical Biology. doi:10.1002/9780470048672.wecb493. ISBN 0470048670.
- 1 2 3 4 5 Francis, M. B.; Carrico, I. S. (2010). "New frontiers in protein bioconjugation". Current Opinion in Chemical Biology. 14 (6): 771–773. doi:10.1016/j.cbpa.2010.11.006. PMID 21112236.
- 1 2 Kalia, J.; Raines, R. T. (2010). "Advances in Bioconjugation". Current Organic Chemistry. 14 (2): 138–147. doi:10.2174/138527210790069839. PMC 2901115. PMID 20622973.
- 1 2 Carrico, I. S.; Carlson, B. L.; Bertozzi, C. R. (2007). "Introducing genetically encoded aldehydes into proteins". Nature Chemical Biology. 3 (6): 321–322. doi:10.1038/nchembio878. PMID 17450134.
- 1 2 Agard, N. J.; Prescher, J. A.; Bertozzi, C. R. (2004). "A Strain-Promoted \3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems". Journal of the American Chemical Society. 126 (46): 15046–15047. doi:10.1021/ja044996f. PMID 15547999.
- ↑ Kolb, H. C.; Finn, M. G.; Sharpless, K. B. (2001). "Click Chemistry: Diverse Chemical Function from a Few Good Reactions". Angewandte Chemie International Edition. 40 (11): 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. PMID 11433435.
- 1 2 3 Rostovtsev, Vsevolod V.; Green, Luke G.; Fokin, Valery V.; Sharpless, K. Barry (2002). "A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective "Ligation" of Azides and Terminal Alkynes". Angewandte Chemie International Edition. 41 (14): 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. ISSN 1433-7851. PMID 12203546.
- ↑ Koniev, O.; Wagner, A. (2015). "Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation". Chem. Soc. Rev. 44 (15): 5495–5551. doi:10.1039/C5CS00048C.
- ↑ Thordarson, P.; Le Droumaguet, B.; Velonia, K. (2006). "Well-defined protein–polymer conjugates—synthesis and potential applications". Applied Microbiology and Biotechnology. 73 (2): 243–254. doi:10.1007/s00253-006-0574-4.
- ↑ Yang,, W.; Thordarson, P. (2007). "Carbon nanotubes for biological and biomedical applications". Nanotechnology. 18 (41).
- ↑ Gerber, HP; Senter, PD; Grewal, IS (2009). "Antibody drug-conjugates targeting the tumor vasculature: Current and future developments". MAbs. 1 (3): 247–53. doi:10.4161/mabs.1.3.8515. PMC 2726597. PMID 20069754. Archived from the original on February 2, 2014.
- ↑ Dovgan, I.; Ursuegui, S.; Erb, S.; Michel, C.; Kolodych, S.; Cianférani, S.; Wagner, A. (2017). "Acyl Fluorides: Fast, Efficient, and Versatile Lysine-Based Protein Conjugation via Plug-and-Play Strategy". Bioconjugate Chem. doi:10.1021/acs.bioconjchem.7b00141.
- ↑ Fodje, M. N.; Al-Karadaghi, S. (2002). "Occurrence, conformational features and amino acid propensities for the π-helix". Protein Eng. 15: 353–358.
- ↑ Shiu, H.-Y.; Chan, T.-C.; Ho, C.-M.; Lin, Y.; Wong, M.-K.; Che, C.-M. (2009). "Electron-Deficient Alkynes as Cleavable Reagents for the Modification of Cysteine-Containing Peptides in Aqueous Medium". Chem. Eur. J. 15 (15): 3839–3850. doi:10.1002/chem.200800669.
- ↑ Koniev, O.; Leriche, G.; Nothisen, M.; Remy, J.-S.; Strub, J.-M.; Schaeffer-Reiss, C.; Dorsselaer, A.; Baati, R.; Wagner, A. (2014). "Selective Irreversible Chemical Tagging of Cysteine with 3-Arylpropiolonitriles". Bioconjugate Chem. 25 (2): 202–206. doi:10.1021/bc400469d.
- ↑ Ban, H.; Nagano, M.; Gavrilyuk, J.; Barbas, C.F. (2015). "Facile and Stabile Linkages through Tyrosine: Bioconjugation Strategies with the Tyrosine-Click Reaction". Bioconjugate Chem. 4 (24): 520–532. doi:10.1021/bc300665t. PMC 3658467.
- ↑ Joshi, N.S.; Whitaker, L.R.; Francis, M.B. (2004). "A Three-Component Mannich-Type Reaction for Selective Tyrosine Bioconjugation". J. Am. Chem. Soc. 126 (49): 15942–15943. doi:10.1021/ja0439017.
- 1 2 Dirksen, A.; Hackeng, T. M.; Dawson, P. E. (2006). "Nucleophilic Catalysis of Oxime Ligation". Angewandte Chemie International Edition. 45 (45): 7581–4. doi:10.1002/anie.200602877. PMID 17051631.
- ↑ Kool, Eric; Park, Do-Hyoung; Crisalli, Pete. "Fast Hydrazone Reactants: Electronic and Acid/Base Effects Strongly Influence Rate at Biological pH". Journal of the American Chemical Society. 135 (47): 17663–17666. doi:10.1021/ja407407h. PMC 3874453. PMID 24224646.
- ↑ Schmidt, Pascal; Zhou, Linna; Tishinov, Kiril; Zimmermann, Kaspar; Gillingham, Dennis. "Dialdehydes Lead to Exceptionally Fast Bioconjugations at Neutral pH by Virtue of a Cyclic Intermediate". Angewandte Chemie International Edition. 53 (41): 10928–10931. doi:10.1002/anie.201406132. PMID 25164607.
- ↑ Schmidt, Pascal; Stress, Cedric; Gillingham, Dennis. "Boronic acids facilitate rapid oxime condensations at neutral pH". Chemical Science. 6: 3329–3333. doi:10.1039/C5SC00921A.
- ↑ Lemieux, G. A.; De Graffenrie, C. L.; Bertozzi, C. R. (2003). "A Fluorogenic Dye Activated by the Staudinger Ligation". Journal of the American Chemical Society. 125 (16): 4708–4709. doi:10.1021/ja029013y. PMID 12696879.
- ↑ Laughlin, S. T.; Baskin, J. M.; Amacher, S. L.; Bertozzi, C. R. (2008). "In Vivo Imaging of Membrane-Associated Glycans in Developing Zebrafish". Science. 320 (5876): 664–667. Bibcode:2008Sci...320..664L. doi:10.1126/science.1155106. PMC 2701225. PMID 18451302.
- ↑ Saxon, E.; Bertozzi, C. R. (2000). "Cell Surface Engineering by a Modified Staudinger Reaction". Science. 287 (5460): 2007–2010. Bibcode:2000Sci...287.2007S. doi:10.1126/science.287.5460.2007. PMID 10720325.
- ↑ Prescher, J. A.; Dube, D. H.; Bertozzi, C. R. (2004). "Chemical remodelling of cell surfaces in living animals". Nature. 430 (7002): 873–877. Bibcode:2004Natur.430..873P. doi:10.1038/nature02791. PMID 15318217.
- ↑ Lin, F. L.; Hoyt, H. M.; Van Halbeek, H.; Bergman, R. G.; Bertozzi, C. R. (2005). "Mechanistic Investigation of the Staudinger Ligation". Journal of the American Chemical Society. 127 (8): 2686–2695. doi:10.1021/ja044461m. PMID 15725026.
- ↑ Chang, P. V.; Prescher, J. A.; Sletten, E. M.; Baskin, J. M.; Miller, I. A.; Agard, N. J.; Lo, A.; Bertozzi, C. R. (2010). "Copper-free click chemistry in living animals". Proceedings of the National Academy of Sciences. 107 (5): 1821. Bibcode:2010PNAS..107.1821C. doi:10.1073/pnas.0911116107. PMC 2836626.