FASTQ format
FASTQ format is a text-based format for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores. Both the sequence letter and quality score are each encoded with a single ASCII character for brevity.
| Developed by | Wellcome Trust Sanger Institute |
|---|---|
| Initial release | ~2000 |
| Type of format | Bioinformatics |
| Extended from | ASCII and FASTA format |
| Website | maq |
It was originally developed at the Wellcome Trust Sanger Institute to bundle a FASTA formatted sequence and its quality data, but has recently become the de facto standard for storing the output of high-throughput sequencing instruments such as the Illumina Genome Analyzer.[1]
Format
A FASTQ file normally uses four lines per sequence.
- Line 1 begins with a '@' character and is followed by a sequence identifier and an optional description (like a FASTA title line).
- Line 2 is the raw sequence letters.
- Line 3 begins with a '+' character and is optionally followed by the same sequence identifier (and any description) again.
- Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
A FASTQ file containing a single sequence might look like this:
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
The byte representing quality runs from 0x21 (lowest quality; '!' in ASCII) to 0x7e (highest quality; '~' in ASCII). Here are the quality value characters in left-to-right increasing order of quality (ASCII):
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
The original Sanger FASTQ files also allowed the sequence and quality strings to be wrapped (split over multiple lines), but this is generally discouraged as it can make parsing complicated due to the unfortunate choice of "@" and "+" as markers (these characters can also occur in the quality string).
Illumina sequence identifiers
Sequences from the Illumina software use a systematic identifier:
@HWUSI-EAS100R:6:73:941:1973#0/1
| HWUSI-EAS100R | the unique instrument name |
|---|---|
| 6 | flowcell lane |
| 73 | tile number within the flowcell lane |
| 941 | 'x'-coordinate of the cluster within the tile |
| 1973 | 'y'-coordinate of the cluster within the tile |
| #0 | index number for a multiplexed sample (0 for no indexing) |
| /1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
Versions of the Illumina pipeline since 1.4 appear to use #NNNNNN instead of #0 for the multiplex ID, where NNNNNN is the sequence of the multiplex tag.
With Casava 1.8 the format of the '@' line has changed:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
| EAS139 | the unique instrument name |
|---|---|
| 136 | the run id |
| FC706VJ | the flowcell id |
| 2 | flowcell lane |
| 2104 | tile number within the flowcell lane |
| 15343 | 'x'-coordinate of the cluster within the tile |
| 197393 | 'y'-coordinate of the cluster within the tile |
| 1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| Y | Y if the read is filtered, N otherwise |
| 18 | 0 when none of the control bits are on, otherwise it is an even number |
| ATCACG | index sequence |
Note that more recent versions of Illumina software output a sample number (as taken from the sample sheet) in place of an index sequence. For example, the following header might appear in the first sample of a batch:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:N:18:1
NCBI Sequence Read Archive
FASTQ files from the INSDC Sequence Read Archive often include a description, e.g.
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC +SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
In this example there is an NCBI-assigned identifier, and the description holds the original identifier from Solexa/Illumina (as described above) plus the read length. Sequencing was performed in paired-end mode (~500bp insert size), see SRR001666. The default output format of fastq-dump produces entire spots, containing any technical reads and typically single or paired-end biological reads. Its behavior was driven by the demands of several projects current at the time fastq-dump was developed, most notably the 1000 Genomes Project.
$ fastq-dump.2.9.0 -Z -X 2 SRR001666
Read 2 spots for SRR001666
Written 2 spots for SRR001666
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACCAAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9ICIIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGAAGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBIIIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
Modern usage of FASTQ almost always involves splitting the spot into its biological reads, as described in submitter-provided metadata:
$ fastq-dump -X 2 SRR001666 --split-3
Read 2 spots for SRR001666
Written 2 spots for SRR001666
$ head SRR001666_1.fastq SRR001666_2.fastq
==> SRR001666_1.fastq <==
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA
+SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI
==> SRR001666_2.fastq <==
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
IIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
When present in the archive, fastq-dump can attempt to restore read names to original format. NCBI does not store original read names by default:
$ fastq-dump -X 2 SRR001666 --split-3 --origfmt
Read 2 spots for SRR001666
Written 2 spots for SRR001666
$ head SRR001666_1.fastq SRR001666_2.fastq
==> SRR001666_1.fastq <==
@071112_SLXA-EAS1_s_7:5:1:817:345
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+071112_SLXA-EAS1_s_7:5:1:817:345
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
@071112_SLXA-EAS1_s_7:5:1:801:338
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA
+071112_SLXA-EAS1_s_7:5:1:801:338
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI
==> SRR001666_2.fastq <==
@071112_SLXA-EAS1_s_7:5:1:817:345
AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+071112_SLXA-EAS1_s_7:5:1:817:345
IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@071112_SLXA-EAS1_s_7:5:1:801:338
AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+071112_SLXA-EAS1_s_7:5:1:801:338
IIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
In the example above, the original read names were used rather than the accessioned read name. NCBI accessions runs and the reads they contain. Original read names, assigned by sequencers, are able to function as locally unique identifiers of a read, and convey exactly as much information as a serial number. The ids above were algorithmically assigned based upon run information and geometric coordinates. Early SRA loaders parsed these ids and stored their decomposed components internally. NCBI stopped recording read names because they are frequently modified from the vendors' original format in order to associate some additional information meaningful to a particular processing pipeline, and this caused name format violations that resulted in a high number of rejected submissions. Without a clear schema for read names, their function remains that of an unique read id, conveying the same amount of information as a read serial number.
Also note that fastq-dump converts this FASTQ data from the original Solexa/Illumina encoding to the Sanger standard (see encodings below). This is because the SRA serves as a repository for NGS information, rather than format. The various *-dump tools are capable of producing data in several formats from the same source. The requirements for doing so have been dictated by users over several years, with the majority of early demand coming from the 1000 Genomes Project.
Variations
Quality
A quality value Q is an integer mapping of p (i.e., the probability that the corresponding base call is incorrect). Two different equations have been in use. The first is the standard Sanger variant to assess reliability of a base call, otherwise known as Phred quality score:

The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used a different mapping, encoding the odds p/(1-p) instead of the probability p:

Although both mappings are asymptotically identical at higher quality values, they differ at lower quality levels (i.e., approximately p > 0.05, or equivalently, Q < 13).
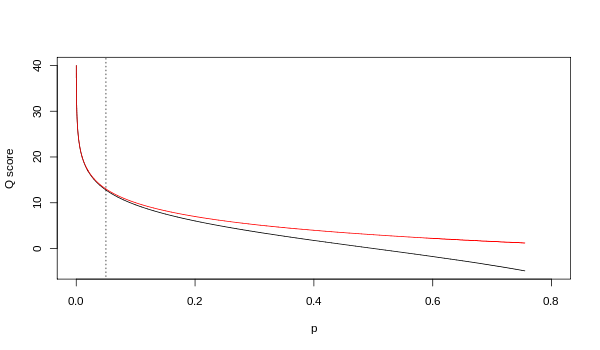
At times there has been disagreement about which mapping Illumina actually uses. The user guide (Appendix B, page 122) for version 1.4 of the Illumina pipeline states that: "The scores are defined as Q=10*log10(p/(1-p)) [sic], where p is the probability of a base call corresponding to the base in question".[2] In retrospect, this entry in the manual appears to have been an error. The user guide (What's New, page 5) for version 1.5 of the Illumina pipeline lists this description instead: "Important Changes in Pipeline v1.3 [sic]. The quality scoring scheme has changed to the Phred [i.e., Sanger] scoring scheme, encoded as an ASCII character by adding 64 to the Phred value. A Phred score of a base is: , where e is the estimated probability of a base being wrong.[3]

Encoding
- Sanger format can encode a Phred quality score from 0 to 93 using ASCII 33 to 126 (although in raw read data the Phred quality score rarely exceeds 60, higher scores are possible in assemblies or read maps). Also used in SAM format.[4] Coming to the end of February 2011, Illumina's newest version (1.8) of their pipeline CASAVA will directly produce fastq in Sanger format, according to the announcement on seqanswers.com forum.[5]
- Solexa/Illumina 1.0 format can encode a Solexa/Illumina quality score from -5 to 62 using ASCII 59 to 126 (although in raw read data Solexa scores from -5 to 40 only are expected)
- Starting with Illumina 1.3 and before Illumina 1.8, the format encoded a Phred quality score from 0 to 62 using ASCII 64 to 126 (although in raw read data Phred scores from 0 to 40 only are expected).
- Starting in Illumina 1.5 and before Illumina 1.8, the Phred scores 0 to 2 have a slightly different meaning. The values 0 and 1 are no longer used and the value 2, encoded by ASCII 66 "B", is used also at the end of reads as a Read Segment Quality Control Indicator.[6] The Illumina manual[7] (page 30) states the following: If a read ends with a segment of mostly low quality (Q15 or below), then all of the quality values in the segment are replaced with a value of 2 (encoded as the letter B in Illumina's text-based encoding of quality scores)... This Q2 indicator does not predict a specific error rate, but rather indicates that a specific final portion of the read should not be used in further analyses. Also, the quality score encoded as "B" letter may occur internally within reads at least as late as pipeline version 1.6, as shown in the following example:
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT +HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
An alternative interpretation of this ASCII encoding has been proposed.[8] Also, in Illumina runs using PhiX controls, the character 'B' was observed to represent an "unknown quality score". The error rate of 'B' reads was roughly 3 phred scores lower the mean observed score of a given run.
- Starting in Illumina 1.8, the quality scores have basically returned to the use of the Sanger format (Phred+33).
For raw reads, the range of scores will depend on the technology and the base caller used, but will typically be up to 41 for recent Illumina chemistry. Since the maximum observed quality score was previously only 40, various scripts and tools break when they encounter data with quality values larger than 40. For processed reads, scores may be even higher. For example, quality values of 45 are observed in reads from Illumina's Long Read Sequencing Service (previously Moleculo).
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS..................................................... ..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX...................... ...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...................... .................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ..................... LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL.................................................... !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ | | | | | | 33 59 64 73 104 126 0........................26...31.......40 -5....0........9.............................40 0........9.............................40 3.....9..............................41 0.2......................26...31........41 S - Sanger Phred+33, raw reads typically (0, 40) X - Solexa Solexa+64, raw reads typically (-5, 40) I - Illumina 1.3+ Phred+64, raw reads typically (0, 40) J - Illumina 1.5+ Phred+64, raw reads typically (3, 41) with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold) (Note: See discussion above). L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
Color space
For SOLiD data, the sequence is in color space, except the first position. The quality values are those of the Sanger format. Alignment tools differ in their preferred version of the quality values: some include a quality score (set to 0, i.e. '!') for the leading nucleotide, others do not. The sequence read archive includes this quality score.
Simulation
FASTQ read simulation has been approached by several tools.[9][10] A comparison of those tools can be seen here.[11]
Compression
General compressors
General-purpose tools such as Gzip and bzip2 regard FASTQ as a plain text file and result in suboptimal compression ratios. NCBI's Sequence Read Archive encodes metadata using the LZ-77 scheme. General FASTQ compressors typically compress distinct fields (read names, sequences, comments, and quality scores) in a FASTQ file separately; these include DSRC and DSRC2, FQC, LFQC, Fqzcomp, and Slimfastq.
Reads
Having a reference genome around is convenient because then instead of storing the nucleotide sequences themselves, one can just align the reads to the reference genome and store the positions (pointers) and mismatches; the pointers can then be sorted according to their order in the reference sequence and encoded, e.g., with run-length encoding. When the coverage or the repeat content of the sequenced genome is high, this leads to a high compression ratio. Unlike the SAM/BAM formats, FASTQ files do not specify a reference genome. Alignment-based FASTQ compressors supports the use of either user-provided or de novo assembled reference: LW-FQZip uses a provided reference genome and Quip, Leon, k-Path and KIC perform de novo assembly using a de Brujin graph-based approach.
Explicit read mapping and de novo assembly are typically slow. Reordering-based FASTQ compressors first cluster reads that share long substrings and then independently compress reads in each cluster after reordering them or assembling them into longer contigs, achieving perhaps the best trade-off between the running time and compression rate. SCALCE is the first such tool, followed by Orcom and Mince. BEETL uses a generalized Burrows–Wheeler transform for reordering reads, and HARC achieves better performance with hash-based reordering. AssemblTrie instead assembles reads into reference trees with as few total number of symbols as possible in the reference.[12][13]
Benchmarks for these tools are available in [14].
Quality values
Quality values account for about half of the required disk space in the FASTQ format (before compression), and therefore the compression of the quality values can significantly reduce storage requirements and speed up analysis and transmission of sequencing data. Both lossless and lossy compression are recently being considered in the literature. For example, the algorithm QualComp [15] performs lossy compression with a rate (number of bits per quality value) specified by the user. Based on rate-distortion theory results, it allocates the number of bits so as to minimize the MSE (mean squared error) between the original (uncompressed) and the reconstructed (after compression) quality values. Other algorithms for compression of quality values include SCALCE [16] and Fastqz.[17] Both are lossless compression algorithms that provide an optional controlled lossy transformation approach. For example, SCALCE reduces the alphabet size based on the observation that “neighboring” quality values are similar in general. For a benchmark, see [18].
As of the HiSeq 2500 Illumina gives the option to output qualities that have been coarse grained into quality bins. The binned scores are computed directly from the empirical quality score table, which is itself tied to the hardware, software and chemistry that were used during the sequencing experiment.[19]
Encryption
The encryption of FASTQ files has been mostly addressed with a specific encryption tool: Cryfa.[20] Cryfa uses AES encryption and enables to compact data besides encryption. It can also address FASTA files.
File extension
There is no standard file extension for a FASTQ file, but .fq and .fastq are commonly used.
Format converters
- Biopython version 1.51 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- EMBOSS version 6.1.0 patch 1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioPerl version 1.6.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioRuby version 1.4.0 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioJava version 1.7.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
See also
References
- Cock, P. J. A.; Fields, C. J.; Goto, N.; Heuer, M. L.; Rice, P. M. (2009). "The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants". Nucleic Acids Research. 38 (6): 1767–1771. doi:10.1093/nar/gkp1137. PMC 2847217. PMID 20015970.
- Sequencing Analysis Software User Guide: For Pipeline Version 1.4 and CASAVA Version 1.0, dated April 2009 PDF Archived June 10, 2010, at the Wayback Machine
- Sequencing Analysis Software User Guide: For Pipeline Version 1.5 and CASAVA Version 1.0, dated August 2009 PDF
- Sequence/Alignment Map format Version 1.0, dated August 2009 PDF
- Seqanswer's topic of skruglyak, dated January 2011 website
- Illumina Quality Scores, Tobias Mann, Bioinformatics, San Diego, Illumina http://seqanswers.com/forums/showthread.php?t=4721
- Using Genome Analyzer Sequencing Control Software, Version 2.6, Catalog # SY-960-2601, Part # 15009921 Rev. A, November 2009 http://watson.nci.nih.gov/solexa/Using_SCSv2.6_15009921_A.pdf%5B%5D
- SolexaQA project website
- Huang, W; Li, L; Myers, J. R.; Marth, G. T. (2012). "ART: A next-generation sequencing read simulator". Bioinformatics. 28 (4): 593–4. doi:10.1093/bioinformatics/btr708. PMC 3278762. PMID 22199392.
- Pratas, D; Pinho, A. J.; Rodrigues, J. M. (2014). "XS: A FASTQ read simulator". BMC Research Notes. 7: 40. doi:10.1186/1756-0500-7-40. PMC 3927261. PMID 24433564.
- Escalona, Merly; Rocha, Sara; Posada, David (2016). "A comparison of tools for the simulation of genomic next-generation sequencing data". Nature Reviews Genetics. 17 (8): 459–69. doi:10.1038/nrg.2016.57. PMC 5224698. PMID 27320129.
- Ginart AA, Hui J, Zhu K, Numanagić I, Courtade TA, Sahinalp SC; et al. (2018). "Optimal compressed representation of high throughput sequence data via light assembly". Nat Commun. 9 (1): 566. doi:10.1038/s41467-017-02480-6. PMC 5805770. PMID 29422526.CS1 maint: multiple names: authors list (link)
- Zhu, Kaiyuan; Numanagić, Ibrahim; Sahinalp, S. Cenk (2018). "Genomic Data Compression". Encyclopedia of Big Data Technologies. Cham: Springer International Publishing. pp. 779–783. doi:10.1007/978-3-319-63962-8_55-1. ISBN 978-3-319-63962-8.
- Numanagić, Ibrahim; Bonfield, James K; Hach, Faraz; Voges, Jan; Ostermann, Jörn; Alberti, Claudio; Mattavelli, Marco; Sahinalp, S Cenk (2016-10-24). "Comparison of high-throughput sequencing data compression tools". Nature Methods. Springer Science and Business Media LLC. 13 (12): 1005–1008. doi:10.1038/nmeth.4037. ISSN 1548-7091.
- Ochoa, Idoia; Asnani, Himanshu; Bharadia, Dinesh; Chowdhury, Mainak; Weissman, Tsachy; Yona, Golan (2013). "Qual Comp: A new lossy compressor for quality scores based on rate distortion theory". BMC Bioinformatics. 14: 187. doi:10.1186/1471-2105-14-187. PMC 3698011. PMID 23758828.
- Hach, F; Numanagic, I; Alkan, C; Sahinalp, S. C. (2012). "SCALCE: Boosting sequence compression algorithms using locally consistent encoding". Bioinformatics. 28 (23): 3051–7. doi:10.1093/bioinformatics/bts593. PMC 3509486. PMID 23047557.
- fastqz.http://mattmahoney.net/dc/fastqz/
- M. Hosseini, D. Pratas, and A. Pinho. 2016. A survey on data compression methods for biological sequences. Information 7(4):(2016): 56
- Illumina Tech Note.http://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/technote_understanding_quality_scores.pdf
- Hosseini M, Pratas D, Pinho A (2018). Cryfa: a secure encryption tool for genomic data. Bioinformatics. 35. pp. 146–148. doi:10.1093/bioinformatics/bty645.
External links
- MAQ webpage discussing FASTQ variants
- Fastx toolkit collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing
- Fastqc quality control tool for high throughput sequence data
- GTO toolkit for FASTQ data
- FastQC Fastqc on a bwHPC-C5 system in Germany
- PRINSEQ can be used for QC and to filter, reformat, or trim sequence data (web-based and command line versions)
- Cryfa can be used for secure encryption of FASTQ, FASTA, VCF and SAM/BAM files (command line version)