Virtual screening
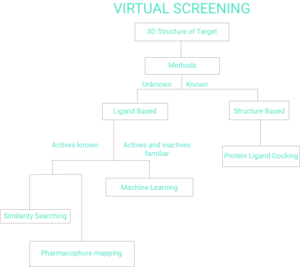
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.[2][3]
Virtual screening has been defined as the "automatically evaluating very large libraries of compounds" using computer programs.[4] As this definition suggests, VS has largely been a numbers game focusing on how the enormous chemical space of over 1060 conceivable compounds[5] can be filtered to a manageable number that can be synthesized, purchased, and tested. Although searching the entire chemical universe may be a theoretically interesting problem, more practical VS scenarios focus on designing and optimizing targeted combinatorial libraries and enriching libraries of available compounds from in-house compound repositories or vendor offerings. As the accuracy of the method has increased, virtual screening has become an integral part of the drug discovery process.[6][1] Virtual Screening can be used to select in house database compounds for screening, choose compounds that can be purchased externally, and to choose which compound should be synthesized next.
Methods
There are two broad categories of screening techniques: ligand-based and structure-based.[7] The remainder of this page will reflect Figure 1 Flow Chart of Virtual Screening.
Ligand-based
Given a set of structurally diverse ligands that binds to a receptor, a model of the receptor can be built by exploiting the collective information contained in such set of ligands. These are known as pharmacophore models. A candidate ligand can then be compared to the pharmacophore model to determine whether it is compatible with it and therefore likely to bind.[8]
Another approach to ligand-based virtual screening is to use 2D chemical similarity analysis methods[9] to scan a database of molecules against one or more active ligand structure.
A popular approach to ligand-based virtual screening is based on searching molecules with shape similar to that of known actives, as such molecules will fit the target's binding site and hence will be likely to bind the target. There are a number of prospective applications of this class of techniques in the literature.[10][11][12] Pharmacophoric extensions of these 3D methods are also freely-available as webservers.[13][14]
Structure-based
Structure-based virtual screening involves docking of candidate ligands into a protein target followed by applying a scoring function to estimate the likelihood that the ligand will bind to the protein with high affinity.[15][16][17] Webservers oriented to prospective virtual screening are available to all.[18][19]
Hybrid methods
Hybrid methods that rely on structural and ligand similarity were also developed to overcome the limitations of traditional VLS approaches [20][21][22] This methodologies utilizes evolution‐based ligand‐binding information to predict small-molecule binders[23][22][24] and can employ both global structural similarity and pocket similarity.[21][23] A global structural similarity based approach employs both an experimental structure or a predicted protein model to find structural similarity with proteins in the PDB holo‐template library. Upon detecting significant structural similarity, 2D fingerprint based Tanimoto coefficient metric is applied to screen for small-molecules that are similar to ligands extracted from selected holo PDB templates.[25][26] The predictions from this method have been experimentally assessed [22] and shows good enrichment in identifying active small molecules.
The above specified method depends on global structural similarity and is not capable of a priori selecting a particular ligand‐binding site in the protein of interest. Further, since the methods rely on 2D similarity assessment for ligands, they are not capable of recognizing stereochemical similarity of small-molecules that are substantially different but demonstrate geometric shape similarity. To address these concerns, a new pocket centric approach, PoLi, capable of targeting specific binding pockets in holo‐protein templates, was developed and experimentally assessed[20][21].
Computing Infrastructure
The computation of pair-wise interactions between atoms, which is a prerequisite for the operation of many virtual screening programs, is of computational complexity, where N is the number of atoms in the system. Because of the quadratic scaling with respect to the number of atoms, the computing infrastructure may vary from a laptop computer for a ligand-based method to a mainframe for a structure-based method.

Ligand-based
Ligand-based methods typically require a fraction of a second for a single structure comparison operation. A single CPU is enough to perform a large screening within hours. However, several comparisons can be made in parallel in order to expedite the processing of a large database of compounds.
Structure-based
The size of the task requires a parallel computing infrastructure, such as a cluster of Linux systems, running a batch queue processor to handle the work, such as Sun Grid Engine or Torque PBS.
A means of handling the input from large compound libraries is needed. This requires a form of compound database that can be queried by the parallel cluster, delivering compounds in parallel to the various compute nodes. Commercial database engines may be too ponderous, and a high speed indexing engine, such as Berkeley DB, may be a better choice. Furthermore, it may not be efficient to run one comparison per job, because the ramp up time of the cluster nodes could easily outstrip the amount of useful work. To work around this, it is necessary to process batches of compounds in each cluster job, aggregating the results into some kind of log file. A secondary process, to mine the log files and extract high scoring candidates, can then be run after the whole experiment has been run.
Accuracy
The aim of virtual screening is to identify molecules of novel chemical structure that bind to the macromolecular target of interest. Thus, success of a virtual screen is defined in terms of finding interesting new scaffolds rather than the total number of hits. Interpretations of virtual screening accuracy should therefore be considered with caution. Low hit rates of interesting scaffolds are clearly preferable over high hit rates of already known scaffolds.
Most tests of virtual screening studies in the literature are retrospective. In these studies, the performance of a VS technique is measured by its ability to retrieve a small set of previously known molecules with affinity to the target of interest (active molecules or just actives) from a library containing a much higher proportion of assumed inactives or decoys. By contrast, in prospective applications of virtual screening, the resulting hits are subjected to experimental confirmation (e.g., IC50 measurements). There is consensus that retrospective benchmarks are not good predictors of prospective performance and consequently only prospective studies constitute conclusive proof of the suitability of a technique for a particular target.[27][28][29][30]
Application to drug discovery
Virtual screening is a very useful application when it comes to identifying hit molecules as a beginning for medicinal chemistry. As the virtual screening approach begins to become a more vital and substantial technique within the medicinal chemistry industry the approach has had an expeditious increase.[31]
Ligand-based methods
While not knowing the structure trying to predict how the ligands will bind to the receptor. With the use of pharmacophore features each ligand identified donor, and acceptors. Equating features are overlaid, however given it is unlikely there is a single correct solution.[1]
Pharmacophore models
This technique is used when merging the results of searches by using unlike reference compounds, same descriptors and coefficient, but different active compounds. This technique is beneficial because it is more efficient than just using a single reference structure along with the most accurate performance when it comes to diverse actives.[1]
Pharmacophore is an ensemble of steric and electronic features that are needed to have an optimal supramolecular interaction or interactions witha biological target structure in order to precipitate it's biological response. Choose a representative as a set of actives, most methods will look for similar bindings. It is preferred to have multiple rigid molecules and the ligands should be diversified, in other words ensure to have different features that don't occur during the binding phase.[1]
Structure
Build a compound predictive model based on known active and known inactive knowledge. QSAR's (Quantitative-Structure Activity Relationship) which is restricted to a small homogenous dataset. SAR's (Structure Activity Relationship) where data is treated qualitatively and can be used with structural classes and more than one binding mode. Models prioritize compounds for lead discovery.[1]
Machine Learning
In order to use Machine Learning for this model of Virtual Screening there must be a training set with known active and known inactive compounds. There also is a model of activity that then is computed by way of substructural analysis, recursive partitioning, support vector machines, k nearest neighbors ad neural networks. The final step is finding the probability that a compound is active and then ranking each compound based on its probability of being active.[1]
Substructural Analysis in Machine Learning
The first Machine Learning model used on large datasets is the Substructure Analysis that was created in 1973. Each fragment substructure make a continuous contribution an activity of specific type.[1] Substructure is a method that overcomes the difficulty of massive dimensionality when it comes to analyzing structures in drug design. An efficient substructure analysis is used for structures that have similarities to a multi-level building or tower. Geometry is used for numbering boundary joints for a given structure in the onset and towards the climax. When the method of special static condensation and substitutions routines are developed this method is proved to be more productive than the previous substructure analysis models.[32]
Recursive Partitioning
Recursively partitioning is method that creates a decision tree using qualitative data. Understanding the way rules break classes up with a low error of misclassification while repeating each step until no sensible splits can be found. However, recursive partitioning can have poor prediction ability potentially creating fine models at the same rate.[1]
Structure Based Methods Known Protein Ligand Docking
Ligand can bind into an active site within a protein by using a docking search algorithm, and scoring function in order to identify the most likely cause for an individual ligand while assigning a priority order.[1]
See also
References
- 1 2 3 4 5 6 7 8 9 10 Gillet V (2013). "Ligand-Based and Structure-Based Virtual Screening" (PDF). The University of Sheffield.
- ↑ Rester U (July 2008). "From virtuality to reality - Virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective". Current Opinion in Drug Discovery & Development. 11 (4): 559–68. PMID 18600572.
- ↑ Rollinger JM, Stuppner H, Langer T (2008). "Virtual screening for the discovery of bioactive natural products". Progress in Drug Research. Progress in Drug Research. 65 (211): 211, 213–49. doi:10.1007/978-3-7643-8117-2_6. ISBN 978-3-7643-8098-4. PMID 18084917.
- ↑ Walters WP, Stahl MT, Murcko MA (1998). "Virtual screening – an overview". Drug Discov. Today. 3 (4): 160–178. doi:10.1016/S1359-6446(97)01163-X.
- ↑ Bohacek RS, McMartin C, Guida WC (1996). "The art and practice of structure-based drug design: a molecular modeling perspective". Med. Res. Rev. 16: 3–50. doi:10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6.
- ↑ McGregor MJ, Luo Z, Jiang X (June 11, 2007). "Chapter 3: Virtual screening in drug discovery". In Huang Z. Drug Discovery Research. New Frontiers in the Post-Genomic Era. Wiley-VCH: Weinheim, Germany. pp. 63–88. ISBN 978-0-471-67200-5.
- ↑ McInnes C (October 2007). "Virtual screening strategies in drug discovery". Current Opinion in Chemical Biology. 11 (5): 494–502. doi:10.1016/j.cbpa.2007.08.033. PMID 17936059.
- ↑ Sun H (2008). "Pharmacophore-based virtual screening". Current Medicinal Chemistry. 15 (10): 1018–24. doi:10.2174/092986708784049630. PMID 18393859.
- ↑ Willet P, Barnard JM, Downs GM (1998). "Chemical similarity searching". J Chem Inf Comput Sci. 38 (6): 983–996. doi:10.1021/ci9800211.
- ↑ Rush TS, Grant JA, Mosyak L, Nicholls A (March 2005). "A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction". Journal of Medicinal Chemistry. 48 (5): 1489–95. doi:10.1021/jm040163o. PMID 15743191.
- ↑ Ballester PJ, Westwood I, Laurieri N, Sim E, Richards WG (February 2010). "Prospective virtual screening with Ultrafast Shape Recognition: the identification of novel inhibitors of arylamine N-acetyltransferases". Journal of the Royal Society, Interface / the Royal Society. 7 (43): 335–42. doi:10.1098/rsif.2009.0170. PMC 2842611. PMID 19586957.
- ↑ Kumar A, Zhang KY (2018). "Advances in the Development of Shape Similarity Methods and Their Application in Drug Discovery". Frontiers in Chemistry. 6: 315. doi:10.3389/fchem.2018.00315. PMID 30090808.
- ↑ Li H, Leung KS, Wong MH, Ballester PJ (July 2016). "USR-VS: a web server for large-scale prospective virtual screening using ultrafast shape recognition techniques". Nucleic Acids Research. 44 (W1): W436–41. doi:10.1093/nar/gkw320. PMID 27106057.
- ↑ Sperandio O, Petitjean M, Tuffery P (July 2009). "wwLigCSRre: a 3D ligand-based server for hit identification and optimization". Nucleic Acids Research. 37 (Web Server issue): W504–9. doi:10.1093/nar/gkp324. PMC 2703967. PMID 19429687.
- ↑ Kroemer RT (August 2007). "Structure-based drug design: docking and scoring". Current Protein & Peptide Science. 8 (4): 312–28. doi:10.2174/138920307781369382. PMID 17696866.
- ↑ Cavasotto CN, Orry AJ (2007). "Ligand docking and structure-based virtual screening in drug discovery". Current Topics in Medicinal Chemistry. 7 (10): 1006–14. doi:10.2174/156802607780906753. PMID 17508934.
- ↑ Kooistra AJ, Vischer HF, McNaught-Flores D, Leurs R, de Esch IJ, de Graaf C (2016). "Function-specific virtual screening for GPCR ligands using a combined scoring method". Scientific Reports. 6: 28288. doi:10.1038/srep28288. PMC 4919634. PMID 27339552.
- ↑ Irwin JJ, Shoichet BK, Mysinger MM, Huang N, Colizzi F, Wassam P, Cao Y (September 2009). "Automated docking screens: a feasibility study". Journal of Medicinal Chemistry. 52 (18): 5712–20. doi:10.1021/jm9006966. PMC 2745826. PMID 19719084.
- ↑ Li H, Leung KS, Ballester PJ, Wong MH (2014-01-24). "istar: a web platform for large-scale protein-ligand docking". PLoS One. 9 (1): e85678. doi:10.1371/journal.pone.0085678. PMC 3901662. PMID 24475049.
- 1 2 Srinivasan B, Tonddast-Navaei S, Roy A, Zhou H, Skolnick J (September 2018). "Chemical space of Escherichia coli dihydrofolate reductase inhibitors: New approaches for discovering novel drugs for old bugs". Medicinal Research Reviews. doi:10.1002/med.21538. PMID 30192413.
- 1 2 3 Roy A, Srinivasan B, Skolnick J (August 2015). "PoLi: A Virtual Screening Pipeline Based on Template Pocket and Ligand Similarity". Journal of Chemical Information and Modeling. 55 (8): 1757–70. doi:10.1021/acs.jcim.5b00232. PMC 4593500. PMID 26225536.
- 1 2 3 Srinivasan B, Zhou H, Kubanek J, Skolnick J (2014-04-26). "Experimental validation of FINDSITE(comb) virtual ligand screening results for eight proteins yields novel nanomolar and micromolar binders". Journal of Cheminformatics. 6 (1): 16. doi:10.1186/1758-2946-6-16. PMC 4038399. PMID 24936211.
- 1 2 Zhou H, Skolnick J (January 2013). "FINDSITE(comb): a threading/structure-based, proteomic-scale virtual ligand screening approach". Journal of Chemical Information and Modeling. 53 (1): 230–40. doi:10.1021/ci300510n. PMC 3557555. PMID 23240691.
- ↑ Roy A, Skolnick J (February 2015). "LIGSIFT: an open-source tool for ligand structural alignment and virtual screening". Bioinformatics. 31 (4): 539–44. doi:10.1093/bioinformatics/btu692. PMC 4325547. PMID 25336501.
- ↑ Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP (January 2012). "ChEMBL: a large-scale bioactivity database for drug discovery". Nucleic Acids Research. 40 (Database issue): D1100–7. doi:10.1093/nar/gkr777. PMC 3245175. PMID 21948594.
- ↑ Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (January 2006). "DrugBank: a comprehensive resource for in silico drug discovery and exploration". Nucleic Acids Research. 34 (Database issue): D668–72. doi:10.1093/nar/gkj067. PMC 1347430. PMID 16381955.
- ↑ Irwin JJ (2008). "Community benchmarks for virtual screening". Journal of Computer-Aided Molecular Design. 22 (3–4): 193–9. doi:10.1007/s10822-008-9189-4. PMID 18273555.
- ↑ Good AC, Oprea TI (2008). "Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection?". Journal of Computer-Aided Molecular Design. 22 (3–4): 169–78. doi:10.1007/s10822-007-9167-2. PMID 18188508.
- ↑ Schneider G (April 2010). "Virtual screening: an endless staircase?". Nature Reviews. Drug Discovery. 9 (4): 273–6. doi:10.1038/nrd3139. PMID 20357802.
- ↑ Ballester PJ (January 2011). "Ultrafast shape recognition: method and applications". Future Medicinal Chemistry. 3 (1): 65–78. doi:10.4155/fmc.10.280. PMID 21428826.
- ↑ Lavecchia A, Di Giovanni C (2013). "Virtual screening strategies in drug discovery: a critical review". Current Medicinal Chemistry. 20 (23): 2839–60. doi:10.2174/09298673113209990001. PMID 23651302.
- ↑ Gurujee CS, Deshpande VL (February 1978). "An improved method of substructure analysis". Computers & Structures. 8 (1): 147–152. doi:10.1016/0045-7949(78)90171-2.
Further reading
- Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (May 2007). "Optimization of biaryl piperidine and 4-amino-2-biarylurea MCH1 receptor antagonists using QSAR modeling, classification techniques and virtual screening". Journal of Computer-Aided Molecular Design. 21 (5): 251–67. doi:10.1007/s10822-007-9112-4. PMID 17377847.
- Afantitis A, Melagraki G, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (February 2006). "Investigation of substituent effect of 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides on CCR5 binding affinity using QSAR and virtual screening techniques". Journal of Computer-Aided Molecular Design. 20 (2): 83–95. doi:10.1007/s10822-006-9038-2. PMID 16783600.
- Eckert H, Bajorath J (March 2007). "Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches". Drug Discovery Today. 12 (5–6): 225–33. doi:10.1016/j.drudis.2007.01.011. PMID 17331887.
- Willett P (December 2006). "Similarity-based virtual screening using 2D fingerprints". Drug Discovery Today. 11 (23–24): 1046–53. doi:10.1016/j.drudis.2006.10.005. PMID 17129822.
- Fara DC, Oprea TI, Prossnitz ER, Bologa CG, Edwards BS, Sklar LA (2006). "Integration of virtual and physical screening". Drug Discovery Today: Technologies. 3 (4): 377–385. doi:10.1016/j.ddtec.2006.11.003.
- Muegge I, Oloffa S (2006). "Advances in virtual screening". Drug Discovery Today: Technologies. 3 (4): 405–411. doi:10.1016/j.ddtec.2006.12.002.
- Schneider G (April 2010). "Virtual screening: an endless staircase?". Nature Reviews. Drug Discovery. 9 (4): 273–6. doi:10.1038/nrd3139. PMID 20357802.
External links
- VLS3D – list of over 2000 databases, online and standalone in silico tools