Top-down proteomics
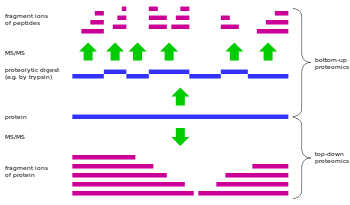
Top-down proteomics is a method of protein identification that uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry analysis.[1][2] Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins.[3] The name is derived from the similar approach to DNA sequencing.[4] Proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance (Penning trap)[5], quadrupole ion trap (Paul trap) or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry.[6] Top-down proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.[7]
Currently the most advanced instrument used to map proteoforms is the Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer. This high-performance instrument is able to give a quantitative analysis of proteoforms, identify them, and deeply characterize them with tandem MS/MS.[8]
Advantages
- The main advantages of the top-down approach include the ability to detect degradation products, sequence variants, and combinations of post-translational modifications.[9]
- In Top-down proteomics, researchers can feed intact full proteins directly into mass spectrometers, and are able to capture the distinct characteristics of each one-of-a-kind proteoform. In addition, Top-down proteomics allows for analysis of an entire protein molecule without digestion and also allows for low mass protein detection.[8]
- Top-down proteomics, when accompanied with polyacrylamide gel electrophoresis, can help to complement the bottom-up proteomic approach. Top-down proteomic methods can assist in exposing large deviations from predictions and has been very successfully pursued by combining Gel Elution Liquid-based Fractionation Entrapment Electrophoresis fractionation, protein precipitation, and reverse phase HPLC with electrospray ionization and MS/MS.[10]
- Characterization of small proteins represents a significant challenge for bottom up proteomics due to the inability to generate sufficient tryptic peptides for analysis. Top-down proteomics allows for low mass protein detection, thus increasing the repertoire of proteins known.[11] While Bottom-up proteomics integrates cleaved products from all proteoforms produced by a gene into a single peptide map of the full-length gene product to tabulate and quantify expressed proteins, a major strength of Top-down proteomics is that it enables researchers to quantitatively track one or more proteoforms from multiple samples and to excise these proteoforms for chemical analysis.[10]
Disadvantages
- In the recent past, the top down approach was relegated to analysis of individual proteins or simple mixtures, while complex mixtures and proteins were analyzed by more established methods such as Bottom-up proteomics. Additionally protein identification and proteoform characterization in the TDP (Top-down proteomics) approach suffer from a dynamic range challenge where the same highly abundant species are repeatedly fragmented.[3]
- Although Top-down proteomics can be operated in relatively high output in order to successfully map proteome coverage at a large level, the rate of identifying new proteins after initial rounds reduces quite sharply.[3]
- Top-down proteomics interrogation can overcome problems for identifying individual proteins, but has not been achieved on a large scale due to a lack of intact protein fractionation methods that are integrated with tandem mass spectrometry.[6]
Research and uses
Study One: Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa
- Researchers performed a study of human proteoforms below 30kDa, used primary IMR90 human fibroblasts containing a Ras function construct that were grown in medium.
- Chose to use Top-Down Proteomics to characterize these proteoforms because it is currently the best method for intact proteins, as I discussed Bottom Up digests the protein and does not do a good job of providing a clear image of distinct intact proteoforms.
- Top Down Proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The Top-down quantitation yielded changes in abundance of 1038 cytoplasmic proteoforms.[3]
Study Two: Combining high-throughput MALDI-TOF mass spectrometry and isoelectric focusing gel electrophoresis for virtual 2D gel-based proteomics
- Researchers used top-down proteomics because could identify the exact proteoforms of intact proteins, rather than the bottom-up approach which gives fragment ions of peptides.
- This study used Virtual 2D gel along with Mass Spectrometry in order to separate protein mixtures. MALDI is a computer software that generates the intact masses of the proteins at each isoelectric point. It started with an image of an IPG-IEF (isoelectric focusing) gel selection that was then analyzed by MALDI.[10]
- Top-down proteomics MALDI-TOF/TOF-MS is more tolerant to impurities; does not require biomarker extraction, purification, and separation; and can be directly applied to intact microorganisms.[12]
See also
References
- ↑ Sze SK, Ge Y, Oh H, McLafferty FW (2002). "Top-down mass spectrometry of a 29-kDa protein for characterization of any posttranslational modification to within one residue". Proc. Natl. Acad. Sci. U.S.A. 99 (4): 1774–9. Bibcode:2002PNAS...99.1774S. doi:10.1073/pnas.251691898. PMC 122269. PMID 11842225.
- ↑ Kelleher NL (2004). "Top-down proteomics". Anal. Chem. 76 (11): 197A–203A. doi:10.1021/ac0415657. PMID 15190879.
- 1 2 3 4 Durbin, Kenneth Robert; Fornelli, Luca; Fellers, Ryan T.; Doubleday, Peter F.; Narita, Masashi; Kelleher, Neil L. (2016). "Quantitation and Identification of Thousands of Human Proteoforms Below 30 kDa". Journal of Proteome Research. 15: 976–982. doi:10.1021/acs.jproteome.5b00997. PMC 4794255. PMID 26795204.
- ↑ Smith CL, Cantor CR (1989). "Evolving strategies for making physical maps of mammalian chromosomes". Genome. 31 (2): 1055–8. doi:10.1139/g89-181. PMID 2698822.
- ↑ Bogdanov B, Smith RD (2005). "Proteomics by FTICR mass spectrometry: top down and bottom up". Mass spectrometry reviews. 24 (2): 168–200. Bibcode:2005MSRv...24..168B. doi:10.1002/mas.20015. PMID 15389855.
- 1 2 Tran, John C.; Zamdborg, Leonid; Ahlf, Dorothy R.; Lee, Ji Eun; Catherman, Adam D.; Durbin, Kenneth R.; Tipton, Jeremiah D.; Vellaichamy, Adaikkalam; Kellie, John F. (2011-12-08). "Mapping intact protein isoforms in discovery mode using top-down proteomics". Nature. 480 (7376): 254–258. Bibcode:2011Natur.480..254T. doi:10.1038/nature10575. ISSN 0028-0836. PMC 3237778. PMID 22037311.
- ↑ Parks, Bryan A.; Jiang, Lihua; Thomas, Paul M.; Wenger, Craig D.; Roth, Michael J.; Boyne, Michael T.; Burke, Patricia V.; Kwast, Kurt E.; Kelleher, Neil L. (2007). "Top-Down Proteomics on a Chromatographic Time Scale Using Linear Ion Trap Fourier Transform Hybrid Mass Spectrometers". Analytical Chemistry. 79 (21): 7984–7991. doi:10.1021/ac070553t. PMC 2361135. PMID 17915963.
- 1 2 Chromatography & Mass Spectrometry Solutions (2015-06-23), Quantitative Top-Down Proteomics & Its Impact on Clinical Research/Basic Biology, retrieved 2016-03-31
- ↑ "Thermo Fisher :: Orbitrap :: Top-Down Proteomics". planetorbitrap.com. Retrieved 2016-02-06.
- 1 2 3 Lohnes, Karen; Quebbemann, Neil R.; Liu, Kate; Kobzeff, Fred; Loo, Joseph A.; Ogorzalek Loo, Rachel R. (2016). "Combining high-throughput MALDI-TOF mass spectrometry and isoelectric focusing gel electrophoresis for virtual 2D gel-based proteomics". Methods. 104: 163–169. doi:10.1016/j.ymeth.2016.01.013.
- ↑ Lorenzatto, Karina R.; Kim, Kyunggon; Ntai, Ioanna; Paludo, Gabriela P.; Camargo de Lima, Jeferson; Thomas, Paul M.; Kelleher, Neil L.; Ferreira, Henrique B. (2015-11-06). "Top Down Proteomics Reveals Mature Proteoforms Expressed in Subcellular Fractions of the Echinococcus granulosus Preadult Stage". Journal of Proteome Research. 14 (11): 4805–4814. doi:10.1021/acs.jproteome.5b00642. ISSN 1535-3907. PMC 4638118. PMID 26465659.
- ↑ Demirev, Plamen A.; Feldman, Andrew B.; Kowalski, Paul; Lin, Jeffrey S. (2005). "Top-Down Proteomics for Rapid Identification of Intact Microorganisms". Analytical Chemistry. 77 (22): 7455–7461. doi:10.1021/ac051419g.
Bibliography
- Borchers CH, Thapar R, Petrotchenko EV, et al. (2006). "Combined top-down and bottom-up proteomics identifies a phosphorylation site in stem-loop-binding proteins that contributes to high-affinity RNA binding". Proc. Natl. Acad. Sci. U.S.A. 103 (9): 3094–9. Bibcode:2006PNAS..103.3094B. doi:10.1073/pnas.0511289103. PMC 1413926. PMID 16492733.
- Han X, Jin M, Breuker K, McLafferty FW (2006). "Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons". Science. 314 (5796): 109–12. Bibcode:2006Sci...314..109H. doi:10.1126/science.1128868. PMID 17023655.
- Whitelegge J, Halgand F, Souda P, Zabrouskov V (2006). "Top-down mass spectrometry of integral membrane proteins". Expert review of proteomics. 3 (6): 585–96. doi:10.1586/14789450.3.6.585. PMID 17181473.
External links
- Top Down Proteomics Overview
- Quantitative Top Down Proteomics Video
- Top Down Proteomics Consortium[1]
- ↑ "Home - Consortium for Top Down Proteomics". www.topdownproteomics.org. Retrieved 2016-04-05.