Tellurophenes
Tellurophenes are the tellurium analogue of thiophenes and selenophenes.[1]
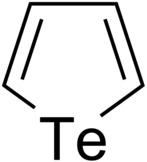
Synthesis
The first preparation of a tellurophene, a tetraphenyltellurophene, was reported in 1961 by Caplier et al.[2] by reacting 1,4-dilithiotetraphenylbutadiene with tellurium tetrachloride, with the former synthesized through reaction of diphenylacetylene and lithium metal. The tellurophene, upon recrystallization from a dichloromethane/ethanol mixture, was obtained in 56% yield, and found to appear as yellow-orange crystals with a melting point of 239-239.5 °C.
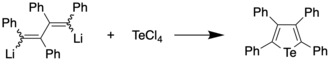
In 1966, Mack was the first to report a synthesis of an unsubstituted tellurophene through the reaction of sodium telluride with diacetylene in methanol at 20 °C.[3] The product was obtained as a pale yellow liquid with a melting and boiling point of -36 °C and 148 °C, respectively. This synthesis was further improved upon by Taticchi et al., who showed that one could obtain higher yields of the product by making sure that there was no moisture and oxygen in the reaction vessel, that pure butadiyne must be used, as it is prone to oxidation and polymerization. Furthermore, the solution must not be concentrated under vacuum to remove the methanol, as this was found to cause loss of the product. This improved procedure allowed the tellurophene to be isolated in 47% yield.
The geometry of tellurophene was first determined in 1973 through microwave spectroscopy, and has been further refined through X-ray diffraction studies.[4] The Te-C bond length is 2.046 Å, longer than that of selenophene and the C-Te-C angle is 82°, smaller than that of selenophene due to tellurium's larger size.
A variety of protocols for the synthesis of tellurophenes have been developed, such as metal-catalyzed cross coupling reactions and cyclization of enynes.[5][6] Some examples are shown below. In 2008, Zeni et al. reported on the copper-catalyzed cyclizations of chalcogenoenynes to obtain 3-substituted chalcogenophenes which could be further functionalized using boronic acids via palladium-catalyzed Suzuki coupling.[5]
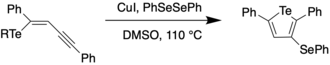
In 2016, Taylor et al. reported a synthetic route to a perfluoroaryl substituted tellurophene through Stille coupling.[6] This compound was then subjected to further iodination and sequential Sonogashira couplings to generate a receptor for anions such as Cl- and Br-.
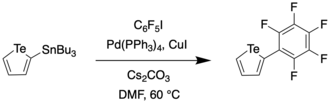
However, metal-catalyzed cross-coupling reactions to synthesize 3-functionalized tellurophenes were deemed to be cumbersome as they required 3-bromo- or 3-iodo-tellurophenes, the syntheses of which could be quite complicated.[7] An alternative method was reported by Seferos et al. in 2013[8], but this method was hindered by low yields and the use of expensive starting materials such as the Weinreb amide.
In 2018, Han et al. reported on a one-pot procedure for the synthesis of a variety of functionalized tellurophenes without the use of transition metals.[7] This was done by reacting substituted 1,1-dibromo-1-en-3-ynes with telluride salts (Na2Te/Na2Se) under mild conditions. The telluride salts were synthesized through an earlier protocol, wherein Te/Se was reduced with sodium borohydride in ethanol.[8] The synthesis of the 3-functionalized tellurophenes is as follows,

It was found through mechanistic studies that the reaction was highly influenced by the polarity of the solvent. Polar solvents such as water were thought to polarize the Te-H bond, thus increasing the negative charge on Te and making it more nucleophilic. To obtain a wider scope of the reaction, the authors used DMF as the solvent since DMF not only has a higher dielectric constant (and therefore, higher polarity) than water, but also was found to be able to dissolve enynes better compared to water. Using a solvent combination of DMF and t-BuOH, the authors were able to synthesize 2,4-disubstituted tellurophenes at room temperature.

Reactivity
Anion receptor
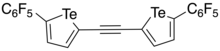
In 2016, Taylor et al. developed a bidentate and electron-deficient bistellurophene receptor in which the tellurophenes were linked through an ethynylene bridge.[6] As the tellurophene was thought to function as a Lewis acid in its interaction with the anion (Lewis base), 2,5-diaryltellurophenes with electron-deficient arene substitutents were synthesized. Through monitoring the change in the optical absorption spectrum upon addition of Bu4N+Cl- in THF, it was found that 2,5-bis[(perfluoro)aryl]tellurophene was able to bind Cl- with an association constant of 310 ± 20 M-1, as well as Br- and BzO-. Using computational studies, it was found that a ethynylene linkage between two tellurophenes would place the chalcogen bond donors at an appropriate distance such that the receptor could form two chalcogen bonds with the chloride. Through sequential Sonogashira coupling reactions, a ethynylene-linked bistellurophene was synthesized from 2-iodo-5-(perfluorophenyl)tellurophene. Upon addition of Bu4N+Cl- to a solution of the receptor in THF, it was found that there were changes to the absorption spectrum, and that the binding association constant (Ka) was 2290 M-1. The significantly higher Ka was found to be in agreement with DFT calculations which showed that the minimum-energy geometry was one where the chloride anion was in between the tellurium atoms, with Te-Cl bond distances of 3.23 Å and Cl-Te-C angles of 170°. One significant difference of the bidentate receptor was that there was no anion-arene stabilizing influence, and operated through purely chalcogen bonding, unlike the monodentate receptor.
Halogen photoelimination
In 2013, Seferos et al. reported the first example of photoreductive elimination of Cl2 and Br2 from an isoindigo-substituted tellurophene, 2,5-bis[5-(N,N′-dihexylisoindigo)]tellurophene.[9] Due to the extensive π-conjugation which resulted in low-energy absorption, relatively low-energy light (505 nm) was used to photoexcite the halogenated species to drive the photoreductive elimination (PE). However, the quantum yields for PE of Cl2 and Br2 were found to be 0.19% and 0.18%, respectively. Through DFT calculations, it was found that the main transition upon photoexcitation was a HOMO to LUMO+2 transition at 535 nm, with the LUMO+2 state possessing Te-X antibonding character. It was postulated that the low quantum yields were due to the fact that there were no lower energy excited states with Te-X antibonding character, and that this would limit the efficiency of the reaction. Therefore, it was thought that by changing the substituents on tellurophene such that the main transition upon photoexcitation would be HOMO to LUMO, this would significantly improve the reaction by removing efficiency losses through relaxations from states that did not possess Te-X antibonding character and did not promote Te-X bond dissociation.
In 2015, Seferos et al. demonstrated that 2,5-diphenyltellurophene (PT) could participate in photoreductive elimination of fluorine, chlorine, and bromine via the two-electron Te(IV)/Te(II) photocycle, with quantum yields of up to 16.9%.[10] This was the first report of an organotellurium compound that could perform photoreductive defluorination. The HOMO and LUMO orbitals of 2,5-diphenyltellurophene were calculated through DFT using the computational program GAMESS, and it shows that the LUMO is delocalized over the entire molecule, in agreement with the orbital pictures reported by Seferos et al.[10] Using DFT calculations using the B3LYP functional, the authors was found that the two strongest optical transitions for PT was the HOMO to LUMO and HOMO to LUMO+1 transitions. Upon addition of halogen, however, it was found that the HOMO-LUMO energy gap decreased, with the LUMO possessing significant Te-X antibonding character. Because of this, it was postulated that by filling the π* orbital with electrons, this would facilitate breaking of the Te-X bond, and hence, halogen dissociation. And indeed, upon addition of excess halogen, the peak at 342 nm corresponding to the tellurophene decreased, while a red-shifted absorption peak appeared, with the peak being more red-shifted as one moved towards the heavier halogens (PT-F2: λmax = 395 nm, PT-Cl2: λmax = 416 nm, PT-Br2: λmax = 433 nm). Upon irradiation of the PT-Br2 sample with a 447.5 nm lamp, it was found that the absorption spectrum of the sample rapidly changed back to that of PT in 12 seconds. This was also observed using 1H NMR spectroscopy.
With F2, however, it was found that there were significant decomposition products owing to the halogen's high reactivity towards PT. This was circumvented by using water as a halogen trap instead of DMBD (2,3-dimethyl-1,3-butadiene), since fluorine exhibits a high reactivity in water to form hydrofluoric acid.
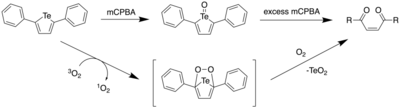
Photooxidation
In 2017, Seferos et al. reported the ring opening of 2,5-diphenyltellurophene (PT) under aerobic conditions, and with mCPBA.[11] Through DFT calculations, it was found that upon oxidation of PT, the resulting Te(IV) oxide PT-O had a lower HOMO level, with the LUMO being significantly stabilized. This led to a large decrease in the HOMO-LUMO energy gap, which predicted a red shift in the maxima of the absorption spectrum. Furthermore, it was found that the electron density of the LUMO included a Te-O σ* orbital.
Addition of 1 equiv of mCPBA to a solution of PT led to an immediate colour change from colourless to yellow. However, upon adding more mCPBA (4 equiv), there was a gradual decrease in the absorbance at 388 nm and a resulting absorption increase below 300 nm. As one would expect a red shift based on computational calculations, it was postulated that upon formation of telluroxide, a different reaction pathway prevented the formation of the tellurone (PT-O2). By analyzing the reaction by NMR spectroscopy, it was found that the yellow solid that had formed as the product had a downfield shift at 1123.3 ppm upon addition of mCPBA. NMR spectra and the absorption spectrum of the product in solution led to the authors attributing this product to the telluroxide.
Optoelectronic properties
In 2018, Okuma et al. reported the synthesis of various 2,5-diaryltellurophenes substituted with electron-donating and electron-withdrawing groups through sequential ditelluride exchange and intramolecular cyclization reactions.[12] By having both electron-donating (e.g. OMe) and electron-withdrawing (e.g. CN) groups on the tellurophene simultaneously, this resulted in a sharp reduction of the HOMO-LUMO gap. Furthermore, the authors observed significant solvatochromism, since the emission maxima shifted to longer wavelengths with increasing solvent polarity. Through DFT calculations, the authors was found that the HOMO was localized on the π-orbitals of the tellurophene and the electron-donating substituent, with the LUMO localized over the π*-orbitals of the tellurophene and the electron-withdrawing substituent. It was concluded that having both electron-donating and electron-withdrawing substituents stabilizes the LUMO, with the HOMO-LUMO transitions having significant charge-transfer character, which in turn explained the solvatochromic effect. This work therefore showed how one can tune the optoelectronic properties of π-conjugated tellurophenes.
The same molecule was subjected to DFT calculations using the computational program GAMESS, where it was found that the HOMO and LUMO orbitals were in qualitative agreement with the orbital pictures reported by Okuma, showing that the HOMO and LUMO show extensive orbital delocalization on the p-anisyl and p-cyanophenyl substituents, respectively.
References
- ↑ Chivers, Tristram; Laitinen, Risto S. (2015). "Tellurium: a maverick among the chalcogens". Chemical Society Reviews. 44 (7): 1725–1739. doi:10.1039/c4cs00434e. ISSN 0306-0012.
- ↑ Braye, E. H.; Hübel, W.; Caplier, I. (1961-11). "New Unsaturated Heterocyclic Systems. I". Journal of the American Chemical Society. 83 (21): 4406–4413. doi:10.1021/ja01482a026. ISSN 0002-7863. Check date values in:
|date=(help) - ↑ Mack, W. "Synthesis of Tellurophene and its 2,5-Disubstituted Derivatives". Angew. Chem. Int. Ed. 5 (10): 896–896.
- ↑ Lukevics, E.; Arsenyan, P.; Belyakov, S.; Pudova, O. (2002). Chemistry of Heterocyclic Compounds. 38 (7): 763–777. doi:10.1023/a:1020607300418. ISSN 0009-3122 http://dx.doi.org/10.1023/a:1020607300418. Missing or empty
|title=(help) - 1 2 Stein, André L.; Alves, Diego; da Rocha, Juliana T.; Nogueira, Cristina W.; Zeni, Gilson (2008-11-06). "Copper Iodide-Catalyzed Cyclization of (Z)-Chalcogenoenynes". Organic Letters. 10 (21): 4983–4986. doi:10.1021/ol802060f. ISSN 1523-7060.
- 1 2 3 4 Garrett, Graham E.; Carrera, Elisa I.; Seferos, Dwight S.; Taylor, Mark S. (2016). "Anion recognition by a bidentate chalcogen bond donor". Chemical Communications. 52 (64): 9881–9884. doi:10.1039/c6cc04818h. ISSN 1359-7345.
- 1 2 Karapala, Vamsi Krishna; Shih, Hong-Pin; Han, Chien-Chung (2018-03). "Cascade and Effective Syntheses of Functionalized Tellurophenes". Organic Letters. 20 (6): 1550–1554. doi:10.1021/acs.orglett.8b00279. ISSN 1523-7060. Check date values in:
|date=(help) - 1 2 Jahnke, Ashlee A.; Djukic, Brandon; McCormick, Theresa M.; Buchaca Domingo, Ester; Hellmann, Christoph; Lee, Yunjeong; Seferos, Dwight S. (2013-01-11). "Poly(3-alkyltellurophene)s Are Solution-Processable Polyheterocycles". Journal of the American Chemical Society. 135 (3): 951–954. doi:10.1021/ja309404j. ISSN 0002-7863.
- ↑ Carrera, Elisa I.; McCormick, Theresa M.; Kapp, Marius J.; Lough, Alan J.; Seferos, Dwight S. (2013-11-19). "Thermal and Photoreductive Elimination from the Tellurium Center of π-Conjugated Tellurophenes". Inorganic Chemistry. 52 (23): 13779–13790. doi:10.1021/ic402485d. ISSN 0020-1669.
- 1 2 3 Carrera, Elisa I.; Seferos, Dwight S. (2015). "Efficient halogen photoelimination from dibromo, dichloro and difluoro tellurophenes". Dalton Transactions. 44 (5): 2092–2096. doi:10.1039/c4dt01751j. ISSN 1477-9226.
- 1 2 Carrera, Elisa I.; Seferos, Dwight S. (2017-05-10). "Ring Opening of π-Delocalized 2,5-Diphenyltellurophene by Chemical or Self-Sensitized Aerobic Photooxidation". Organometallics. 36 (14): 2612–2621. doi:10.1021/acs.organomet.7b00240. ISSN 0276-7333.
- ↑ Nagahora, Noriyoshi; Yahata, Shuhei; Goto, Shoko; Shioji, Kosei; Okuma, Kentaro (2018-02-02). "2,5-Diaryltellurophenes: Effect of Electron-Donating and Electron-Withdrawing Groups on their Optoelectronic Properties". The Journal of Organic Chemistry. 83 (4): 1969–1975. doi:10.1021/acs.joc.7b02906. ISSN 0022-3263.