Letterer–Siwe disease
| Letterer–Siwe disease | |
|---|---|
| Synonyms | Acute and disseminated Langerhans cell histiocytosis |
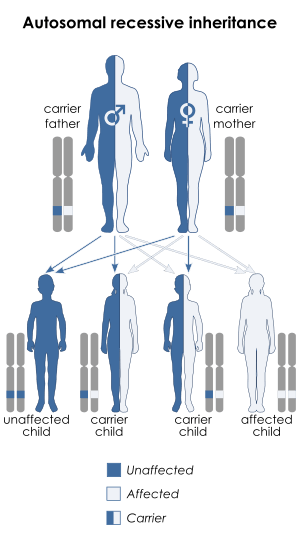 | |
| This condition is inherited in an autosomal recessive manner | |
| Specialty |
Oncology |
Letterer–Siwe disease is one of the four recognized clinical syndromes of Langerhans cell histiocytosis (LCH). It causes approximately 10% of LCH disease and is the most severe form.[1] Prevalence is estimated at 1:500,000 and the disease almost exclusively occurs in children less than three years old.[2] The name is derived from the names of Erich Letterer and Sture Siwe.
Clinical Presentation
Letterer-Siwe is characterized by skin lesions, ear drainage, lymphadenopathy, osteolytic lesions, and hepatosplenomegaly. The skin lesions are scaly and may involve the scalp, ear canals, and abdomen.[3]
Cause
Oncogenic mutation of BRAF 50-70% cases
Prognosis
The disease is often rapidly fatal, with a five year survival rate of 50%. The development of thrombocytopenia is a poor prognostic sign.[1]
References
- 1 2 "Langerhans Cell Histiocytosis - Hematology and Oncology - Merck Manuals Professional Edition". Merck Manuals Professional Edition. Retrieved 2017-05-19.
- ↑ RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Letterer Siwe disease". www.orpha.net. Retrieved 2017-05-19.
- ↑ "Langerhans cell histiocytosis | DermNet New Zealand". www.dermnetnz.org. Retrieved 2017-05-19.
External links
| Classification | |
|---|---|
| External resources |