Organostannane addition
Organostannane addition reactions comprise the nucleophilic addition of an allyl-, allenyl-, or propargylstannane to an aldehyde, imine, or, in rare cases, a ketone.[1]
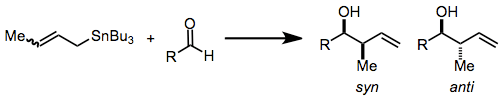
Organostannane addition to carbonyl groups constitutes one of the most common and efficient methods for the construction of contiguous, oxygen-containing stereocenters in organic molecules. As many molecules containing this motif—polypropionates and polyacetates, for instance—are desired by natural products chemists, the title reaction has become important synthetically and has been heavily studied over the years.[2][3] Substituted allylstannanes may create one or two new stereocenters, often with a very high degree of stereocontrol.
Advantages: Organostannanes are known for their stability, ease of handling, and selective reactivity. Chiral allylstannanes often react with great stereoselectivity to give single diastereomers, and models explaining the sense of selectivity are reliable.
Disadvantages: Stoichiometric amounts of metal-containing byproducts are generated during the reaction. Additions to sterically encumbered pi bonds, such as those of ketones, are uncommon.
Mechanism and Stereochemistry
Prevailing Mechanism
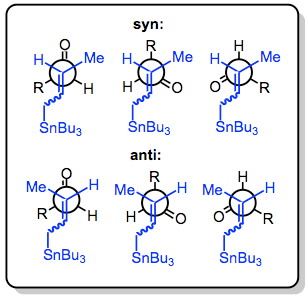
There are three modes for the addition of allylstannanes to carbonyls: thermal addition, Lewis-acid-promoted addition, and addition involving prior transmetalation. Each of these modes invokes a unique model for stereocontrol, but in all cases, a distinction is made between reagent and substrate control. Substrate-controlled additions typically involve chiral aldehydes or imines and invoke the Felkin-Anh model. When all reagents are achiral, only simple diastereoselectivity (syn versus anti, see above) must be considered. Addition takes place via an SE' mechanism involving concerted dissociation of tin and C-C bond formation at the γ position.
With the allylstannane and aldehyde in high-temperature conditions, addition proceeds through a six-membered, cyclic transition state, with the tin center serving as an organizing element. The configuration of the double bond in the allylstannane controls the sense of diastereoselectivity of the reaction.
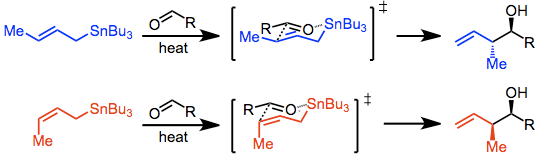
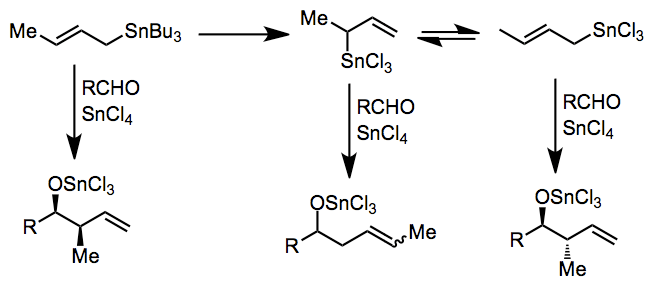
This is not the case in Lewis-acid-promoted reactions, in which either the (Z) or (E) stannane gives the syn product predominantly. The origin of this selectivity has been debated,[4] and depends on the relative energies of a number of acyclic, rotameric transition states. (E)-stannanes exhibit higher syn selectivity than the corresponding (Z) stannanes.
In the presence of particularly strong Lewis acids, transmetalation may occur before addition. Complex reaction mixtures may result if transmetalation is not complete or if an equilibrium between allylic isomers exists. Tin(IV) chloride[5] and indium(III) chloride[6] have been employed for useful reactions in this mode.
Enantioselective Variants
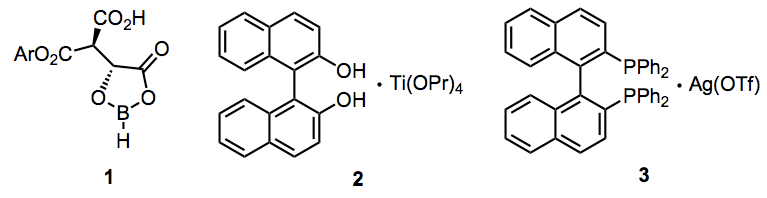
A wide variety of enantioselective additions employing chiral, non-racemic Lewis acids are known. The chiral (acyloxy)borane or "CAB" catalyst 1, titanium-BINOL system 2, and silver-BINAP system 3 provide addition products in high ee via the Lewis-acid-promoted mechanism described above.
Scope and Limitations
The possibility of incorporating oxygen-containing substituents into allyl- and allenylstannanes expands their scope and utility substantially over methods relying on more promiscuous organometallics. These compounds are usually prepared by enantioselective reduction with a chiral reducing agent such as BINAL-H.[7] In the presence of only a Lewis acid, isomerization of α-alkoxy allylstannanes to the corresponding γ-alkoxy isomers can occur.
The use of chiral electrophiles is common, and can provide "double stereocontrol" if the stannane is also chiral. Chelation control using Lewis acids such as magnesium bromide can lead to high stereoselectivities for reactions of α-alkoxy aldehydes.
Nucleophilic addition into propargyl mesylates or tosylates is used to form allenylstannanes.[8] These compounds react similarly to allylstannanes, affording homopropargyl alcohols.
Imines are less reactive than the corresponding aldehydes, but palladium catalysis can be used to facilitate addition into imines. The use of iminium ions as electrophiles has also been reported.[9]
Synthetic Applications
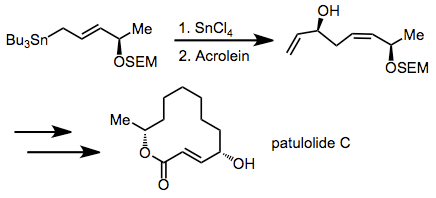
In an interesting application of the transmetalation methodology, the chiral allylstannane shown added to acrolein to yield the 1,5-syn diastereomer as a single stereoisomer. A subsequent sigmatropic rearrangement increased the distance between the stereocenters even further. This step was carried out en route to (±)-Patulolide C.[10]
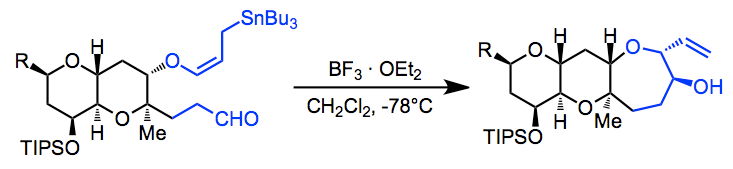
Repeated use of the allylstannane addition in an intramolecular sense was used in the synthesis of Hemibrevetoxin B (one example is shown below). The pseudoequatorial positions of both "appendages" in the starting material lead to the observed stereochemistry.[11]
Comparison with Other Methods
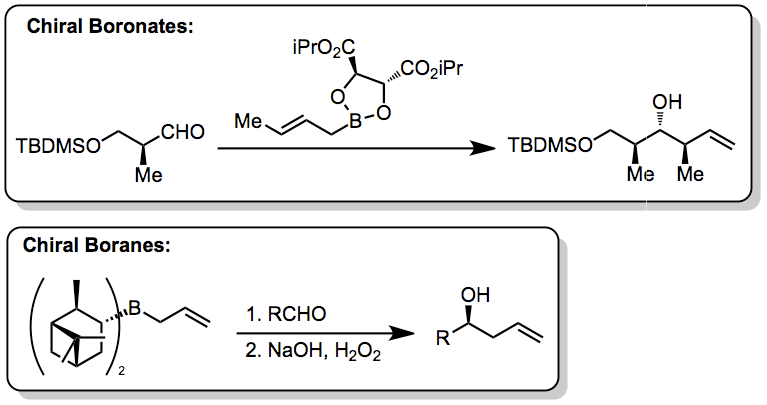
Methods for the addition of allyl groups to carbonyl compounds are numerous and incorporate a variety of metals. Organochromium,[12] organozinc,[13] and organoindium[14] reagents have been used in additions to carbonyls. Allylsilane reagents are more hydrolytically stable and less toxic than allyltin compounds, but are also less reactive. Catalytic, enantioselective versions of allylsilane additions are similar to those discussed here and rely on chiral Lewis acids.[15] Allylboronate[16] and -borane[17] reagents have also been developed for enantioselective addition to carbonyls—in this class of reactions, the allylboron reagent confers stereochemical control.
Experimental Conditions and Procedure
Typical Conditions
Additions in the absence of a catalyst or promoter take place only upon heating to 100-130 °C. In the presence of a Lewis acid, the addition will proceed at -78 °C. Reactions must be carried out under an inert atmosphere in anhydrous conditions, but the process is operationally simple and does not require a great deal of special care. Reactions should be carried out in a well-ventilated fume hood, however, as volatile organotin compounds are highly toxic.
References
- ↑ Gung, B.W. Org. React. 2004, 64, 1-112.
- ↑ Denmark, S. E.; Weber, E. J. J. Am. Chem. Soc. 1984, 106, 7970.
- ↑ Keck, G. E.; Dougherty, S. M.; Savin, K. A. J. Am. Chem. Soc. 1995, 117, 6210.
- ↑ Keck, G. E.; Savin, K. A.; Cressman, E. N. K.; Abbott, D. E. J. Org. Chem. 1994, 59, 7889.
- ↑ McNeill, A. H.; Thomas, E. J. Synthesis 1994, 322.
- ↑ Marshall, J. A.; Hinkle, K. W. J. Org. Chem. 1995, 60, 1920.
- ↑ Marshall, J. A.; Jablonowski, J. A.; Jiang, H. J. Org. Chem. 1999, 64, 2152.
- ↑ Ruitenberg, K.; Vermeer, P. Tetrahedron Lett. 1984, 25, 3019.
- ↑ Yamamoto, Y.; Schmid, M. J. Chem. Soc., Chem. Commun. 1989, 1310.
- ↑ Dorling, E.K.; Thomas, E.J. Tetrahedron Lett. 1999, 40, 471.
- ↑ Kadota, I.; Yamamoto, Y. J. Org. Chem. 1998, 63, 6597.
- ↑ Cintas, P. Synthesis 1992, 248.
- ↑ Jones, P.; Knochel, P. J. Org. Chem. 1999, 64, 186.
- ↑ Marshall, J. A.; Hinkle, K. W. J. Org. Chem. 1996, 61, 105.
- ↑ Ishihara, K.; Mouri, M.; Gao, Q.; Maruyama, T.; Furuta, K.; Yamamoto, H. J. Am. Chem. Soc. 1993, 115, 11490.
- ↑ Roush, W. R.; Hoong, L. K.; Palmer, M. A. J.; Straub, J. A.; Palkowitz, A. D. J. Org. Chem. 1990, 55, 4117.
- ↑ Brown, H. C.; Jadhav, P. K. J. Am. Chem. Soc. 1983, 105, 2092.