Kelvin equation
The Kelvin equation describes the change in vapour pressure due to a curved liquid–vapor interface, such as the surface of a droplet. The vapor pressure at a convex curved surface is higher than that at a flat surface. The Kelvin equation is dependent upon thermodynamic principles and does not allude to special properties of materials. It is also used for determination of pore size distribution of a porous medium using adsorption porosimetry. The equation is named in honor of William Thomson, also known as Lord Kelvin.
Formulation
The Kelvin equation may be written in the form

where is the actual vapour pressure, is the saturated vapour pressure, is the surface tension, is the molar volume of the liquid, is the universal gas constant, is the radius of the droplet, and is temperature.







Equilibrium vapor pressure depends on droplet size.
- If the curvature is convex, then, ,
- If the curvature is concave, then , (where is the vapor pressure when the surface is flat)


As increases, decreases, and the droplets grow into bulk liquid.
If we now cool the vapour, then decreases, but so does . This means increases as the liquid is cooled. We can treat and as approximately fixed, which means that the critical radius must also decrease. The further a vapour is supercooled, the smaller the critical radius becomes. Ultimately it gets as small as a few molecules, and the liquid undergoes homogeneous nucleation and growth.

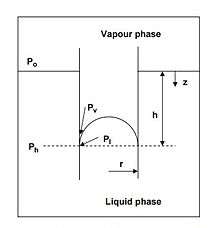
The change in vapor pressure can be attributed to changes in the Laplace pressure. When the Laplace pressure rises in a droplet, the droplet tends to evaporate more easily.
When applying the Kelvin equation, two cases must be distinguished: A drop of liquid in its own vapor will result in a positively curved liquid surface, and a bubble of vapor in a liquid will result in a negatively curved liquid surface.
History
The form of the Kelvin equation here is not the form in which it appeared in Lord Kelvin's article of 1871. The derivation of the form that appears in this article from Kelvin's original equation was presented by Robert von Helmholtz (son of German physicist Hermann von Helmholtz) in his dissertation of 1885.[1]
Apparent paradox
An equation similar to that of Kelvin can be derived for the solubility of small particles or droplets in a liquid, by means of the connection between vapour pressure and solubility, thus the Kelvin equation also applies to solids, to slightly soluble liquids, and their solutions if the partial pressure is replaced by the solubility of the solid (or a second liquid) at the given radius, , and by the solubility at a plane surface. Hence small particles (like small droplets) are more soluble than larger ones.
These results led to the problem of how new phases can ever arise from old ones. For example, if a container filled with water vapour at slightly below the saturation pressure is suddenly cooled, perhaps by adiabatic expansion, as in a cloud chamber, the vapour may become supersaturated with respect to liquid water. It is then in a metastable state, and we may expect condensation to take place. A reasonable molecular model of condensation would seem to be that two or three molecules of water vapour come together to form a tiny droplet, and that this nucleus of condensation then grows by accretion, as additional vapour molecules happen to hit it. The Kelvin equation, however, indicates that a tiny droplet like this nucleus, being only a few ångströms in diameter, would have a vapour pressure many times that of the bulk liquid. As far as tiny nuclei are concerned, the vapour would not be supersaturated at all. Such nuclei should immediately re-evaporate, and the emergence of a new phase at the equilibrium pressure, or even moderately above it should be impossible. Hence, the over-saturation must be several times higher than the normal saturation value for spontaneous nucleation to occur.
There are two ways of resolving this paradox. In the first place, we know the statistical basis of the second law of thermodynamics. In any system at equilibrium, there are always fluctuations around the equilibrium condition, and if the system contains few molecules, these fluctuations may be relatively large. There is always a chance that an appropriate fluctuation may lead to the formation of a nucleus of a new phase, even though the tiny nucleus could be called thermodynamically unstable. The chance of a fluctuation is e−ΔS/k, where ΔS is the deviation of the entropy from the equilibrium value.
It is unlikely, however, that new phases often arise by this fluctuation mechanism and the resultant spontaneous nucleation. Calculations show that the chance, e−ΔS/k, is usually too small. It is more likely that tiny dust particles act as nuclei in supersaturated vapours or solutions. In the cloud chamber, it is the clusters of ions caused by a passing high-energy particle that acts as nucleation centers. Actually, vapours seem to be much less finicky than solutions about the sort of nuclei required. This is because a liquid will condense on almost any surface, but crystallization requires the presence of crystal faces of the proper kind.
See also
References
- ↑ Robert von Helmholtz (1886) "Untersuchungen über Dämpfe und Nebel, besonders über solche von Lösungen" (Investigations of vapors and mists, especially of such things from solutions), Annalen der Physik, 263 (4): 508–543. On pages 523–525, Robert von Helmholtz converts Kelvin's equation to the form that appears here (which is actually the Ostwald–Freundlich equation).
Further reading
- Sir William Thomson (1871) "On the equilibrium of vapour at a curved surface of liquid", Philosophical Magazine, series 4, 42 (282): 448–452.
- W. J. Moore, Physical Chemistry, 4th ed., Prentice Hall, Englewood Cliffs, N. J., (1962) p. 734–736.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, 2nd edition, Academic Press, New York, (1982) p. 121.
- Arthur W. Adamson and Alice P. Gast, Physical Chemistry of Surfaces, 6th edition, Wiley-Blackwell (1997) p. 54.
- Butt, Hans-Jürgen, Karlheinz Graf, and Michael Kappl. "The Kelvin Equation". Physics and Chemistry of Interfaces. Weinheim: Wiley-VCH, 2006. 16–19. Print.
- Anton A. Valeev,"Simple Kelvin Equation Applicable in the Critical Point Vicinity",European Journal of Natural History, (2014), Issue 5, p. 13-14.