Hydrophosphination
Hydrophosphination is the addition of a phosphorus-hydrogen bond across a carbon-carbon multiple bond (Scheme 1) forming a new phosphorus-carbon bond.[1] Hydrophosphination has a high atom economy.[2]
Like other metal-catalyzed heterofunctionalizations, addition of an E-H bond to an unsaturated substrate, a problem is encountered when E is a donor (hydroamination, hydration, hydrothiolation) because the product phosphines can poison the catalyst. Another consideration is that the P-H addition to a carbon-carbon multiple bond can occur in a variety of different ways.[2] The selectivity of the addition is influenced by the catalyst.
Background
A variety of routes exist for the synthesis of phosphines without a catalyst, e.g. using radical, thermal, photochemical.[3][4]
Radical-catalyzed hydrophosphination

Ultraviolet irradiation or conventional free-radical sources initiate radical hydrophosphinations. H-atom abstraction from P-H bond produces the phosphino radical, a seven electron species (Scheme 2).[5] Phosphines with more than one P-H bond can react more than once to form the tertiary phosphines.
Hydrophosphination catalysis
The steps proposed for metal-catalyzed hydrophosphinations typically include coordination of the primary or secondary phosphine to the metal, activation of the P-H bond (via oxidative addition or proton abstraction by an external base), P-C bond formation (through either insertion, Michael addition, or [2+2] cycloaddition), reductive elimination or addition of the abstracted proton and finally substitution of the newly made phosphine by the starting primary or secondary phosphine.[6]
Early transition metal and lanthanide catalysts


Most early metal hydrophosphination catalysts are electron-deficient, d0 metal complexes. Catalysis typically involves inner-sphere P-C bond forming through metal-phosphido intermediates due to the nucleophilicity of the phosphido, being polarized by the metal.[7]
Hydrophosphination of simple alkenes and alkynes is catalyzed by lanthanocene complexes.[8][9][10][11][12] The catalytic cycle for the hydrophosphination of α,ω-pentenylphosphine is shown in Scheme 3. Typical of most electron-poor early metal catalysts for hydrophosphination, this one includes the characteristic P-H bond cleavage and P-C and C-H bond formation steps. The primary phosphine undergoes a σ-bond metathesis with the bis(trimethylsilyl)methylene ligand forming the lanthanide-phosphido. This is then followed by a 1,2-insertion of the pendant terminal alkene or alkyne on the phosphine into the Ln-P bond. Finally, protonolysis of the Ln-C bond with the starting primary phosphine releases the new phosphine and regenerates the catalyst. Given that the metal is electron-poor, the M-C bond is polar enough to be protonolyzed by the substrate primary phosphine; this is characteristic of electron-poor, early metals.
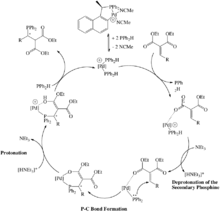
Since many of the transition metal catalysts that are involved in hydrophosphination employ a phosphido intermediate, it is surprising that there are so few examples of catalysts that involve the metal-phosphinidene intermediate, M=PR. One such example is the Ti-catalyzed hydrophosphination of diphenylacetylene with phenylphosphine (Scheme 4) by Mindiola et al.[13] This system involves a cationic catalyst precursor that is stabilized by the bulky 2,4,6-tri(isopropyl)phenyl- substituent on the phosphinidene and the close ionic association of methyltris(pentafluorophenyl)borate. This precursor undergoes exchange with phenylphosphine to make the titanium-phenylphosphinidene complex which is the catalyst. The Ti=PPh undergoes a [2+2] cycloaddition with diphenylacetylene to make the corresponding metallacyclobutene. The substrate, phenylphosphine, protonolyzes the Ti-C bond and after a proton shift regenerates the catalyst and releases the new phosphine.
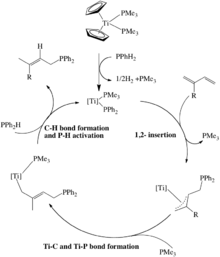
Titanium-catalyzed 1,4-hydrophosphination of 1,3-dienes with diphenylphosphine has been demonstrated (Scheme 5).[14] It is a rare example of a non-d0 early transition metal catalyst that catalyzes hydrophosphination. In the first step, the Ti2+ precursor undergoes an oxidative addition of the P-H bond in diphenylphosphine which generates the Ti3+ catalyst. The rest of the catalysis involve the usual steps: P-C bond formation via 1,2-insertion followed by C-H bond formation and P-H bond activation via protonolysis with the substrate secondary phosphine.
Late transition metal catalysts
Late transition metal hydrophosphination catalysts generally require alkenes and alkynes with electron withdrawing substituents.[15][16][17][18] This is due to the fact that mechanism for the P-C bond forming step usually involves nucleophilic attack of the phosphorus to the unsaturated carbon. Generally speaking, these metal complexes will first be bound by a primary or secondary phosphine, which then undergoes proton abstraction to make a nucleophilic phosphido ligand. The proton abstraction usually occurs by an external base.This base acts as a co-catalyst which is common to late transition metal hydrophosphination catalysts.[19][20][21][22] This complex catalyzes hydrophosphination of enones (Scheme 6). The bound secondary phosphine becomes deprotonated by the base triethylamine, forming the phosphido. Following this the phosphido attacks the β-carbon of the enone, which is bound to the palladium through its oxygen atom. The abstracted proton from the secondary phosphine is delivered to the enone oxygen. Substitution by another secondary phosphine regenerates the catalyst and releases the 1,2-addition product. Other common characteristics of such late metal catalysts that do not require oxidative addition of the P-H bond are: the metal in a 2+ oxidation state, and cationic complexes. These two features are important given that a base is needed because being cationic and in an oxidation state of 2+ increases the acidity of the coordinated primary or secondary phosphine proton and allows for weaker bases/conjugate acids to be used in catalysis.
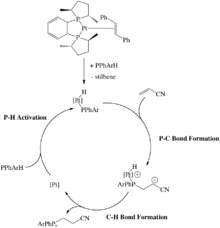
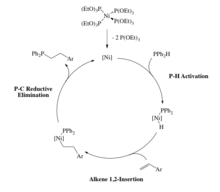
Some late metal hydrophosphination catalysts rely on oxidative addition of a P-H bond. For example, a Pt(0) catalyst that undergoes oxidative addition of a secondary phosphine to form the corresponding Pt(II) phosphido complex (Scheme 7). Following this, alkenes such as acrylonitrile inserts into the Pt-P bond and then reductively eliminate to afford the hydrophosphinated acrylonitrile. This P-C bond forming step was found to occur through an outer-sphere, Michael-type addition because telomerization was occurring. The carbanion intermediate would attack another molecule of acrylonitrile affording a product that would be expected from many 1,2-insertions of the alkene.[23][24][25]
As stated in the last example, the alkene inserted into the metal-phosphorus bond, but insertion into the metal-hydrogen bond is also possible (Scheme 7).[26][27] The Ni(0) catalyst involves oxidation addition of a P-H bond to the metal, followed by insertion of the alkene into the M-H bond (Scheme 8). As a result of this, the product phosphine is produced through reductive elimination of a P-C bond rather than a P-H bond in Glueck's system.

A prospective hydrophosphination catalyst utilizes a ruthenium(II) complex (Scheme 9).[28][29][30][31][32] Like the other late metal catalysts that require a base co-catalyst, this complex requires a base to deprotonate the coordinated secondary phosphine, generating a nucleophilic phosphido complex. The following reactivity varies from the other late metal catalysts in that P-C bond forming step involves a [2+2] cycloaddition with alkenes and alkynes forming metallacycles. The Ru-C bond of the metallacycle is then protonolyzed by the conjugate acid of the base co-catalyst. The newly formed phosphine is then substituted by the starting secondary phosphine to regenerate the catalyst and release the new phosphine. What makes this catalyst unique is that it forms metallacycles, or rather P-C bonds, with strongly and mildly activated, simple and electron-rich alkenes and alkynes, whereas the other catalysts discussed are rather limited; generally, early metals catalyzing unactivated substrates and late metals predominantly activated ones.
As stated, characteristic of the late metal catalysts for hydrophosphination that require a base co-catalyst is that they are in the 2+ oxidation state and cationic. Thus, efforts are also underway to use the similar halide-free, cationic ruthenium(II) complex for catalysis.
References
- ↑ Pullarkat, S. A.; Leung, P. Top. Organomet. Chem. 2013, 43, 145.
- 1 2 Beletskaya, I. P; Ananikov, V. P; Khemchyan, L. L. Catal. Met. Complexes 2013, 37, 213.
- ↑ Rosenberg, L. R. ACS Catal. 2013, 3, 2845.Rosenberg, L. (2013). "Mechanisms of Metal-Catalyzed Hydrophosphination of Alkenes and Alkynes". ACS Catalysis. 3 (12): 2845. doi:10.1021/cs400685c.
- ↑ Pietro Tundo; Liang-Nian He; Ekaterina Lokteva; Claudio Mota (4 October 2016). Chemistry Beyond Chlorine. Springer International Publishing. pp. 152–164. ISBN 978-3-319-30071-9.
- ↑ Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000; pp 28-29.
- ↑ Glueck, D. S. Top. Organomet. Chem. 2010, 31, 65.
- ↑ Greenberg, S.; Stephan, D. W. (2008). "Stoichiometric and catalytic activation of P–H and P–P bonds". Chemical Society Reviews. 37 (8): 1482. doi:10.1039/B612306F.
- ↑ Douglass, M. R.; Stern, C. L.; Marks, T. J. (2001). "Intramolecular Hydrophosphination/Cyclization of Phosphinoalkenes and Phosphinoalkynes Catalyzed by Organolanthanides: Scope, Selectivity, and Mechanism". Journal of the American Chemical Society. 123 (42): 10221. doi:10.1021/ja010811i.
- ↑ Douglass, M. R.; Marks, T. J. J. Am. Chem. Soc. 2000, 122, 1824Douglass, M. R.; Marks, T. J. (2000). "Organolanthanide-Catalyzed Intramolecular Hydrophosphination/Cyclization of Phosphinoalkenes and Phosphinoalkynes". Journal of the American Chemical Society. 122 (8): 1824. doi:10.1021/ja993633q.
- ↑ Douglass, M. R.; Ogasawara, M.; Hong, S.; Metz, M. V.; Marks, T. J. Organometallics, 2002, 21, 283Douglass, M. R.; Ogasawara, M.; Hong, S.; Metz, M. V.; Marks, T. J. (2002). ""Widening the Roof": Synthesis and Characterization of New ChiralC1-Symmetric Octahydrofluorenyl Organolanthanide Catalysts and Their Implementation in the Stereoselective Cyclizations of Aminoalkenes and Phosphinoalkenes". Organometallics. 21 (2): 283. doi:10.1021/om0104013.
- ↑ Kawaoka, A. M.; Douglass, M .R.; Marks, T. J. Organometallics 2003, 22, 4630.Kawaoka, A. M.; Douglass, M. R.; Marks, T. J. (2003). "Homoleptic Lanthanide Alkyl and Amide Precatalysts Efficiently Mediate Intramolecular Hydrophosphination/Cyclization. Observations on Scope and Mechanism". Organometallics. 22 (23): 4630. doi:10.1021/om030439a.
- ↑ Motta, A.; Fragala, I. L.; Marks, T. J. Organometallics, 2005, 24, 4995.Motta, A.; Fragalà, I. L.; Marks, T. J. (2005). "Energetics and Mechanism of Organolanthanide-Mediated Phosphinoalkene Hydrophosphination/Cyclization. A Density Functional Theory Analysis". Organometallics. 24 (21): 4995. doi:10.1021/om050570d.
- ↑ Zhao, G. Y.; Basuli, F.; Kilgore, U. J.; Fan, H. J.; Aneetha, H.; Huffman, J. C.; Wu, G.; Mindiola, D. J. J. Am. Chem. Soc. 2006, 128, 13575.Zhao, G.; Basuli, F.; Kilgore, U. J.; Fan, H.; Aneetha, H.; Huffman, J. C.; Wu, G.; Mindiola, D. J. (2006). "Neutral and Zwitterionic Low-Coordinate Titanium Complexes Bearing the Terminal Phosphinidene Functionality. Structural, Spectroscopic, Theoretical, and Catalytic Studies Addressing the Ti−P Multiple Bond". Journal of the American Chemical Society. 128 (41): 13575. doi:10.1021/ja064853o.
- ↑ Perrier, A.; Comte, V.; Moise, C.; Le Gendre, P. Chem. – Eur. J. 2010, 16, 64.Perrier, A.; Comte, V.; Moïse, C.; Le Gendre, P. (2010). "First Titanium-Catalyzed 1,4-Hydrophosphination of 1,3-Dienes". Chemistry – A European Journal. 16: 64. doi:10.1002/chem.200901863.
- ↑ Pringle, P. G.; Smith, M. B. (1990). "Platinum(0)-catalysed hydrophosphination of acrylonitrile". Journal of the Chemical Society, Chemical Communications (23): 1701. doi:10.1039/C39900001701.
- ↑ Kovacik, I.; Wicht, D. K.; Grewal, N. S.; Glueck, D. S.; Incarvito, C. D.; Guzei, I. A.; Rheingold, A. L. (2000). "Pt(Me-Duphos)-Catalyzed Asymmetric Hydrophosphination of Activated Olefins: Enantioselective Synthesis of Chiral Phosphines". Organometallics. 19 (6): 950. doi:10.1021/om990882e.
- ↑ Sadow, A. D.; Togni, A. J. Am. Chem. Soc. 2005, 127, 17012.Sadow, A. D.; Togni, A. (2005). "Enantioselective Addition of Secondary Phosphines to Methacrylonitrile: Catalysis and Mechanism". Journal of the American Chemical Society. 127 (48): 17012. doi:10.1021/ja0555163.
- ↑ Huang, Y. H.; Pullarkat, S. A.; Li, Y. X.; Leung, P. H. Chem. Commun. 2010, 46, 6950.Huang, Y.; Pullarkat, S. A.; Li, Y.; Leung, P. H. (2010). "Palladium(ii)-catalyzed asymmetric hydrophosphination of enones: Efficient access to chiral tertiary phosphines". Chemical Communications. 46 (37): 6950. doi:10.1039/C0CC00925C. PMID 20730193.
- ↑ Xu, C.; Jun Hao Kennard, G.; Hennersdorf, F.; Li, Y.; Pullarkat, S. A.; Leung, P. H. (2012). "Chiral Phosphapalladacycles as Efficient Catalysts for the Asymmetric Hydrophosphination of Substituted Methylidenemalonate Esters: Direct Access to Functionalized Tertiary Chiral Phosphines". Organometallics. 31 (8): 3022. doi:10.1021/om201115n.
- ↑ Huang, Y. H.; Pullarkat, S. A.; Teong, S.; Chew, R. J.; Li, Y. X.; Leung, P. H. Organometallics 2012, 31, 4871.Huang, Y.; Pullarkat, S. A.; Teong, S.; Chew, R. J.; Li, Y.; Leung, P. H. (2012). "Palladacycle-Catalyzed Asymmetric Intermolecular Construction of Chiral Tertiary P-Heterocycles by Stepwise Addition of H–P–H Bonds to Bis(enones)". Organometallics. 31 (13): 4871. doi:10.1021/om300405h.
- ↑ Huang, Y. H.; Pullarkat, S. A.; Li, Y. X.; Leung, P. H. Inorg. Chem. 2012, 51, 2533.Huang, Y.; Pullarkat, S. A.; Li, Y.; Leung, P. H. (2012). "Palladacycle-Catalyzed Asymmetric Hydrophosphination of Enones for Synthesis of C*- and P*-Chiral Tertiary Phosphines". Inorganic Chemistry. 51 (4): 2533. doi:10.1021/ic202472f. PMID 22289417.
- ↑ Huang, Y.; Chew, R. J.; Li, Y.; Pullarkat, S. A.; Leung, P. H. (2011). "Direct Synthesis of Chiral Tertiary Diphosphinesvia Pd(II)-Catalyzed Asymmetric Hydrophosphination of Dienones". Organic Letters. 13 (21): 5862. doi:10.1021/ol202480r. PMID 21985055.
- ↑ Scriban, C.; Glueck, D. S.; Zakharov, L. N.; Kassel, W. S.; DiPasquale, A. G.; Golen, J. A.; Rheingold, A. L. Organometallics 2006, 25, 5757.Scriban, C.; Glueck, D. S.; Zakharov, L. N.; Kassel, W. S.; Dipasquale, A. G.; Golen, J. A.; Rheingold, A. L. (2006). "P−C and C−C Bond Formation by Michael Addition in Platinum-Catalyzed Hydrophosphination and in the Stoichiometric Reactions of Platinum Phosphido Complexes with Activated Alkenes". Organometallics. 25 (24): 5757. doi:10.1021/om060631n.
- ↑ Scriban, c.; Kovacik, L.; Glueck, D. S. Organometallics, 2005, 24, 4871.Scriban, C.; Kovacik, I.; Glueck, D. S. (2005). "A Protic Additive Suppresses Formation of Byproducts in Platinum-Catalyzed Hydrophosphination of Activated Olefins. Evidence for P−C and C−C Bond Formation by Michael Addition". Organometallics. 24 (21): 4871. doi:10.1021/om050433g.
- ↑ Wicht, D. K.; Kourkine, I. V.; Lew, B. M.; Nthenge, J. M.; Glueck, D. S. J. Am. Chem. Soc. 1997, 119, 5039.Wicht, D. K.; Kourkine, I. V.; Lew, B. M.; Nthenge, J. M.; Glueck, D. S. (1997). "Platinum-Catalyzed Acrylonitrile Hydrophosphination via Olefin Insertion into a Pt−P Bond". Journal of the American Chemical Society. 119 (21): 5039. doi:10.1021/ja970355r.
- ↑ Shulyupin, M. O.; Kazankova, M. A.; Beletskaya, I. P. Org. Lett. 2002, 4, 761.Shulyupin, M. O.; Kazankova, M. A.; Beletskaya, I. P. (2002). "Catalytic Hydrophosphination of Styrenes". Organic Letters. 4 (5): 761. doi:10.1021/ol017238s.
- ↑ Kazankova, M. A.; Shulyupin, M. O.; Borisenko, A. A.; Beletskaya, I. P. Russ. J. Org. Chem. 2002, 38, 1479.Kazankova, M. A.; Shulyupin, M. O.; Borisenko, A. A.; Beletskaya, I. P. (2002). "Synthesis of Alkyl(diphenyl)phosphines by Hydrophosphination of Vinylarenes Catalyzed by Transition Metal Complexes". Russian Journal of Organic Chemistry. 38 (10): 1479. doi:10.1023/A:1022552404812.
- ↑ Derrah, E. J.; Pantazis, D. A.; McDonald, R.; Rosenberg, L. Organometallics 2007, 26, 1473.Derrah, E. J.; Pantazis, D. A.; McDonald, R.; Rosenberg, L. (2007). "A Highly Reactive Ruthenium Phosphido Complex Exhibiting Ru−P π-Bonding". Organometallics. 26 (6): 1473. doi:10.1021/om0700056.
- ↑ Derrah, E. J.; Pantazis, D. A.; McDonald, R.; Rosenberg, L. Angew. Chem. Int. Ed. 2010, 49, 3367.Derrah, E. J.; Pantazis, D. A.; McDonald, R.; Rosenberg, L. (2010). "Concerted [2+2] Cycloaddition of Alkenes to a Ruthenium-Phosphorus Double Bond". Angewandte Chemie International Edition. 49 (19): 3367. doi:10.1002/anie.201000356.
- ↑ Derrah, E. J.; McDonald, R.; Rosenberg, L. Chem. Commun. 2010, 46, 4592.Derrah, E. J.; McDonald, R.; Rosenberg, L. (2010). "The [2+2] cycloaddition of alkynes at a Ru–P π-bond". Chemical Communications. 46 (25): 4592. doi:10.1039/C002765K.
- ↑ Gibson, G. L.; Morrow, K. M. E.; McDonald, R.; Rosenberg, L. Inorg. Chim. Acta. 2011, 369, 133.Gibson, G. L.; Morrow, K. M. E.; McDonald, R.; Rosenberg, L. (2011). "Diastereoselective synthesis of a "chiral-at-Ru" secondary phosphine complex". Inorganica Chimica Acta. 369: 133. doi:10.1016/j.ica.2010.12.058.
- ↑ Burton (née Morrow), K. M. E.; Belli, R. G.; Pantazis, D. A.; McDonald, R.; Rosenberg, L. manuscript in preparation.