Diphosphenes
Diphosphene is a kind of organophosphorus compound that has a phosphorus-phosphorus double bond, denoted by R-P=P-R'. These compounds are not common but are of theoretical interest. Normally, compounds with the empirical formula RP exist as rings. However, like other multiple bonds between heavy main-group elements, P=P double bonds can be stabilized by a large steric hindrance from the substitutions.[1] The first isolated diphosphene bis(2,4,6-tri-tert-butylphenyl)diphosphene was exemplified by Masaaki Yoshifuji and his coworkers in 1981, in which diphosphene is stabilized by two bulky phenyl group.[2]
Synthesis
Synthesis of Aryl-Substituted Diphosphene
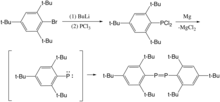
In 1877, Köhler and Michaelis claimed that they synthesized the first isolated diphosphene (PhP=PPh).[4] However, the molecular weight determination[5] and X-ray crystallographic analysis[6] later proved that this "diphosphene" only had a P-P single bond. Then the research to diphosphenes kept silent over almost 20 years until Masaaki Yoshifuji and his coworkers isolated the unprecedented diphosphene--bis(2,4,6-tri-tert-butylphenyl)diphosphene in 1981.[2] They first synthesized the (2,4,6-tri-tert-butylphenyl)phosphorus dichloride by adding phosphorus trichloride to (2,4,6-tri-butylphenyl)lithium that was the product of the lithium and halogen exchange. The phosphorus dichloride dimerized to a diphosphene after magnesium extracted two chlorine from (2,4,6-tri-tert-butylphenyl)phophorus dichloride. P-P bond distance here was measured as 2.034 Å, which was much shorter than the average bond length in (C6H5P)5 (2.217Å) and (C6H5P)6 (2.237Å), indicating its double bond character. This research was a milestone in diphosphene studies because the product here was the first reported compound that had the isolated localized P=P bond. Moreover, this bulky structure provided a instructive pathway for the future synthesis of diphosphene.
Synthesis of Alkyl-Substituted Diphosphene
Tris(trimethylsilyl)methyl group is also a very bulky group that is often used to stabilize multiple bond between heavy elements. By dropwise addition of (Si(Me)3)3CPCl2 to sodium napthelenide. 31P, 1H and 13C spectras all proved the formulation of this alkyl-group stabilized diphosphene.[7]
Synthesis of Boryl-Substituted Diphosphene
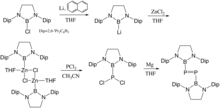
Boryl substituents reveal the potential as both π-electron acceptors and σ-electron donors. The vacant p orbitals enable it to accept the electrons, while low electronegativity reflects the σ donors properties. Computational studies predicted the existence of the boryl-substituted diphosphene and Makoto Yamashita et al. proved it experimentally in 2016. A borylzinc chloride was prepared by from a bulky boryl-lithium compound. This nucleophilic borylzinc compound could attack the phosphorus trichloride and formed boryl-substituted phosphorus dichloride. Similar to the synthesis procedure of a aryl-substituted diphosphene, the boryl-substituted diphosphene was obtained by mixing the boryl-substituted phosphorus dichloride with magnesium. Cyclic voltammogram and UV/Vis Spectrum illustrated that this boryl-substituted diphosphene has lower LUMO level and larger the HOMO-LUMO gap than the aryl-substituted diphosphene.[9]
Structures and Bondings
Experimental Data
X-ray analysis indicates certain important bond lengths and angles of the first diphosphene--bis(2,4,6-tri-tert-butylphenyl)diphosphene: P-P=2.034 Å; P-C=1.826 Å; P-P-C=102.8o; C-P-P-C=172.2o.[3] Compared with the bond length of P-P single bond in H2PPH2 (2.238 Å)[10], the P-P bond distance is much shorter, which reveals double bond character. The P-P double bond in bis(2,4,6-tri-tert-butylphenyl)diphosphene exhibits E configuration. But by visible irradiation of the trans isomer, an interconversion between cis isomer and trans isomers would occur. In 1984, M. Koening et al. noticed a different splitting mode and chemical shift in 1H NMR and 31P NMR under a direct irradiation of a toluene solution of E-bis(2,4,6-tri-tert-butylphenyl)diphosphene, which suggesting a cis-trans isomerization.[11]

Spectroscopic Properties
Diphosphene compounds usually exhibit a symmetry-allowed ( ) (intense) and symmetry-forbidden electronic transitions ( ) (weak).[12] In Raman, there is significant enhancement of P=P stretch in the resonance with allowed electron transition than with the forbidden transition due to different geometries of excited states and enhancement mechanism.[13] Also the observed strong Raman shifts for (CH(SiMe3)2)2P2 and (CH(SiMe3)2P=PC(SiMe3)2) suggest stronger dipnictenes feature of diphosphene compared with P-P single bond.


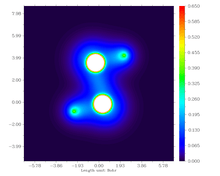
Natural Bond Orbital (NBO) Analysis
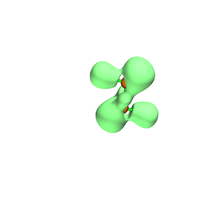
The parent diphosphene (H2P2) has a well localized P=P bond with bond order of 2.03, while all other unbulky substituted diphosphenes have lower bond order. Bond orders of the diphosphenes in which substitutes have extra lone-pair are even lower, due to delocalization of lone pair electrons into P-P . NBO analysis indicates that the bond order of P-P in NaO-P=P-ONa is only 1.62.[14] NBO analysis is also applied in predicting the conformers of original diphosphenes and their properties. H. C. Marsmann's group first introduced halogens into diphosphenes. NBO calculation indicates that C(SiMe3)2SiCl2FP=PC(SiMe3)2SiCl2F has two conformers, one of which contains interactions.[15]


Atoms in Molecules (AIM) Analysis
AIM analysis of bis(2,4,6-tri-tert-butylphenyl)diphosphene under M06-2X/6-31G* level is consistent with the observed crystal data at 300 K. Calculated bond lengths between P-P(2.035 Å) and P-C (1.870 Å) are very close to the observed data. Four atoms in C-P-P-C are not strictly planar but with a slight distortion. Time dependent DFT calculation predicts two absorption peaks at 481 nm and 330 nm, which are raised from HOMO (-6.75 eV) LUMO (-1.28eV) and HOMO-1 (-7.24eV) LUMO. Laplacian map illustrates that the C-P-P-C atoms are correlated covalently. Laplacian of the charge density ( ) at bond critical point between P-P bond is -0.187 while the bond ellipticity is 0.459, both of which indicate that P-P has double bond character. At the BCP between P-C, the value (-0.238) and extremely small ellipticity (0.083) exhibit that the conjugation between P and aromatic substituents is very weak.[16]


Excited Triplet Diphosphenes
Efforts to elucidate the excited states of diphosphenes is important and valueable to realize the application of PP double bonds in molecular electronics. In triplets trans-HPPH, the P-P bond length is predicted to be 2.291 Å. It's not only longer than the P-P double bond in ground state trans-bis(2,4,6-tri-tert-butylphenyl)diphosphene, but also longer than that of P-P single bond in H2PPH2. Calculation of the dihedral angle of trans-HPPH suggests that it's almost 90 degree, which means the formation of and P-P bonds is forbidden and σ bond is enhanced.[17]

Reactivity
Selected reactivity of diphosphenes is summarized in the following picture, including oxidation, reduction, sulfurization, polymerization, carbene addition, halogenation, photolysis and coordination to the transition-metal
Carbene Involved Reactions

Carbene Addition
Similar to the ring-formation behavior in the carbene addition reaction of C=C double bonds, diphosphene can form a P-C-P three-membered ring with dihalocarbene or . Diphosphiranes can further rearrange to 1,3-diphospha-allene via ring opening reactions by using MeLi or n-BuLi.[18]


Carbene Mediated P=P double bond Cleavage
Cleavage of C=C double bonds is very common and important in organic chemistry while that of unsaturated bonds between heavy Group 14 and Group 15 elements are lack of investigation. Successful polarization of Si-Si multiple bonds reveals potential interconversion between -bonding electrons and lone-pair electrons in heavy Group 14 and Group 15 compounds that contain multiple bonds. Diphosphene, as the typical heavy-element multiple bonds in those compounds, can be cleaved by N-heterocyclic carbene (NHC), forming NHC-bound phosphinidenes.[19]
Coordination to Transition-metal
Diphosphenes can bind to transition metal either in a η1 mode by donating a lone pair on phosphorus, or in a η2 behavior via a interaction. If the bulky groups are aryl- groups, arene-coordinated products of η6-type coordination are also possible.
η1-Type Complexes
In 1983, Philip P. Power synthesized a transition-metal complex containing P=P double bond (trans-{[Fe(CO)4][PCH(SiMe3)2]2}) via a simple one-step procedure.[20] They mixed Na2[Fe(CO)4] and dichlorobis(trimethylsilyl)methylphosphine and got a dark red-brown crystals, which was the first complex that contained unbridged P-P double bond. Each phosphorus exhibited terminal coordination nature and the P-P distance was essentially unchanged. Later in 1983, A. H. Cowley reported ArP=PArFe(CO)5(Ar=2,4,6-tri-tert-butylphenyl) by treating diphosephene with Fe2(CO)9 or Na2Fe(CO)4.[21] In this synthesis procedure, there was only one terminal P-coordination and P-P double bond had Z configuration. Apart from iron, other similar transition metal complexes by reacting diphosphenes with transition metal carbonyls of nickel, tungsten and chromium were discovered and they all exhibited Z configuration. M. Yoshifuji proved E/Z isomerization can take place under lighting, probably via migration of the metal moiety from one side to the other.
η2-Type Complexes
Apart from the very bulky substituents, a η2-coordination of diphosphene to a metal is also possible to stabilize the P-P double bond. In 1982, K. R. Dixon et al. synthesized Platinum and Palladium complexes (M(PhP=PPh)L2), {M=Pt, Pd; L=(PPh3)2 or Ph2P[CH2]2PPh2}) which contained side-on coordination.[22] Different from η1 coordination complex, where P-P still kept the double bond nature, P-P distance in side-on coordination complexes (2.121Å in Pd(PhP=PPh)PPh3CH2CH2PPh3) was significantly longer than that in non-coordinated bis(2,4,6-tri-tert-butylphenyl)diphosphene.
η6-Type Complexes
If there are aryl- groups on phosphorus, transition-metal can not only bind to the phosphorus directly, but also form arene-coordinated products of η6-type coordination. Refluxing diphosphene in 1,4-dioxane with the excess of Cr(CO)6 can generate mono and bis arene tricarbonylchromium(0) complexes.[23]
Oxidation
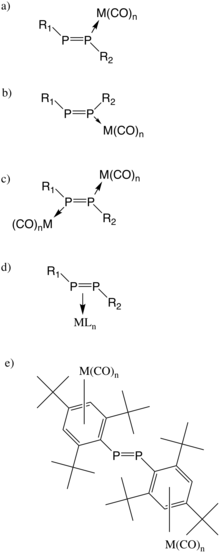
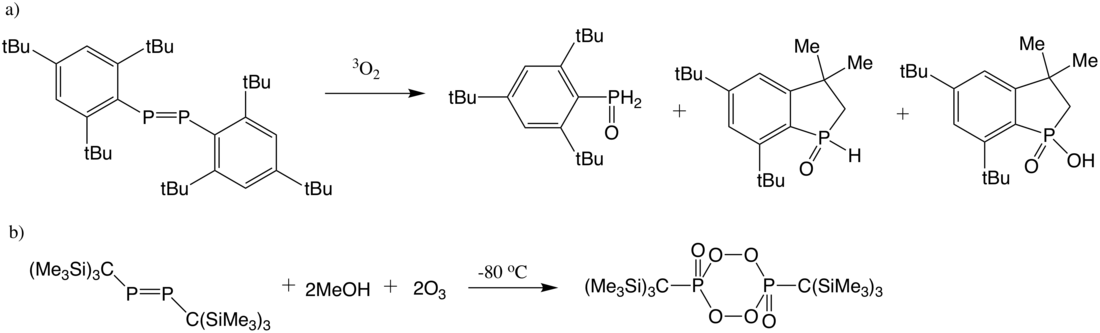
Diphosphene is inert to ground-state oxygen but can be oxidized by triplet oxygen to give a mixture of phosphine oxides and hydroxy benzophosphole oxide.[24] Compared to oxygen involved oxidation, reaction of diphosphene with ozone is much more rapid and indicates a 2:1 (ozone : diphosphene) stoichiometry. Ozonolysis of bis[tris(trimethylsilyl)methyl]diphosphene (Tsi2P2) gives a cyclic diperoxides.[25]
Reduction
Aluminum hydrides (AlH) such as lithium aluminum hydride can reduce diphosphene to give stable diphosphanes: (Ar=2,4,6-tBu3C6H2)[26]

See also
References
- ↑ Power, Philip P. (2010-01-14). "Main-group elements as transition metals". Nature. 463 (7278): 171–177. Bibcode:2010Natur.463..171P. doi:10.1038/nature08634. ISSN 1476-4687.
- 1 2 Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. (1981-07-01). "Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene: isolation of a true phosphobenzene". Journal of the American Chemical Society. 103 (15): 4587–4589. doi:10.1021/ja00405a054. ISSN 0002-7863.
- 1 2 Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. (1981-07-01). "Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene: isolation of a true phosphobenzene". Journal of the American Chemical Society. 103 (15): 4587–4589. doi:10.1021/ja00405a054. ISSN 0002-7863.
- ↑ Kohler, H; Michaelis, A (1877). Ber. Dtsch. Chem. Ges. 10: 807. Missing or empty
|title=(help) - ↑ Kuchen, W; Grilnewald, W (1965). Chem. Ber. 98: 480. Missing or empty
|title=(help) - ↑ Daly, J; Maier, L (1964). Nature. 203: 1167. Bibcode:1964Natur.203.1167D. doi:10.1038/2031167b0. Missing or empty
|title=(help) - ↑ Cowley, A. H.; Kilduff, J. E.; Newman, T. H.; Pakulski, M. (1982-10-01). "Diphosphenes (RP:PR). Synthesis and NMR characterization". Journal of the American Chemical Society. 104 (21): 5820–5821. doi:10.1021/ja00385a060. ISSN 0002-7863.
- ↑ Asami, Shun-suke; Okamoto, Masafumi; Suzuki, Katsunori; Yamashita, Makoto (2016-10-04). "A Boryl-Substituted Diphosphene: Synthesis, Structure, and Reaction with n-Butyllithium To Form a Stabilized Adduct by pπ-pπ Interaction". Angewandte Chemie. 128 (41): 13019–13023. doi:10.1002/ange.201607995. ISSN 1521-3757.
- ↑ Asami, Shun-suke; Okamoto, Masafumi; Suzuki, Katsunori; Yamashita, Makoto (2016-10-04). "A Boryl-Substituted Diphosphene: Synthesis, Structure, and Reaction with n-Butyllithium To Form a Stabilized Adduct by pπ-pπ Interaction". Angewandte Chemie. 128 (41): 13019–13023. doi:10.1002/ange.201607995. ISSN 1521-3757.
- ↑ Matus, Myrna H.; Nguyen, Minh Tho; Dixon, David A. (2007-03-01). "Heats of Formation of Diphosphene, Phosphinophosphinidene, Diphosphine, and Their Methyl Derivatives, and Mechanism of the Borane-Assisted Hydrogen Release". The Journal of Physical Chemistry A. 111 (9): 1726–1736. Bibcode:2007JPCA..111.1726M. doi:10.1021/jp067892v. ISSN 1089-5639.
- ↑ Caminade, Anne-Marie; Verrier, Martine; Ades, Claude; Paillous, Nicole; Koenig, Max (1984-01-01). "Laser irradiation of a diphosphene: evidence for the first cis–trans isomerization". J. Chem. Soc., Chem. Commun. 0 (13): 875–877. doi:10.1039/c39840000875. ISSN 0022-4936.
- ↑ Sasamori, Takahiro; Tokitoh, Norihiro (2008-03-05). "Doubly bonded systems between heavier Group 15 elements". Dalton Trans. 0 (11): 1395–1408. doi:10.1039/b715033d. ISSN 1477-9234.
- ↑ Copeland, Tiffany; Shea, Michael P.; Milliken, Matt C.; Smith, Rhett C.; Protasiewicz, John D.; Simpson, M.Cather. "Raman excitation profile of a sterically protected diphosphene [ArP=PAr]". Analytica Chimica Acta. 496 (1–2): 155–163. doi:10.1016/s0003-2670(03)00996-6.
- ↑ Vogt-Geisse, Stefan; Schaefer, Henry F. (2012-05-08). "Reducing and Reversing the Diphosphene–Diphosphinylidene Energy Separation". Journal of Chemical Theory and Computation. 8 (5): 1663–1670. doi:10.1021/ct300221e. ISSN 1549-9618.
- ↑ Mitrofan, Cristina; Gust, Thorsten; Zanin, Andreas; Bîrzoi, Roxana M.; Jones, Peter G.; du Mont, Wolf-W.; Hayashi, Satoko; Nakanishi, Waro; Marsmann, Heinrich C. (2017-03-17). "Dichlorosilylene Transfer to a P-Fluorophosphaalkene: The Route to a C-Dichlorofluorosilyl-Functionalized Dialkyldiphosphene". European Journal of Inorganic Chemistry. 2017 (11): 1526–1536. doi:10.1002/ejic.201601411. ISSN 1099-0682.
- ↑ Yoshifuji, Masaaki (2017-03-01). "Sterically protected organophosphorus compounds of unusual structures". Pure and Applied Chemistry. 89 (3). doi:10.1515/pac-2016-1029. ISSN 1365-3075.
- ↑ Lu, Tongxiang; Hao, Qiang; Simmonett, Andrew C.; Evangelista, Francesco A.; Yamaguchi, Yukio; Fang, De-Cai; Schaefer, Henry F. (2010-10-14). "Low-Lying Triplet States of Diphosphene and Diphosphinylidene". The Journal of Physical Chemistry A. 114 (40): 10850–10856. Bibcode:2010JPCA..11410850L. doi:10.1021/jp105281w. ISSN 1089-5639.
- 1 2 Yoshifuji, Masaaki; Sasaki, Shigeru; Niitsu, Takashi; Inamoto, Naoki. "A convenient new route from diphosphene to 1,3-diphospha-allene and dynamic NMR studies of the 2,4,6-tri-t-butylphenyl derivative". Tetrahedron Letters. 30 (2): 187–188. doi:10.1016/s0040-4039(00)95155-4.
- 1 2 Hayakawa, Naoki; Sadamori, Kazuya; Tsujimoto, Shota; Hatanaka, Miho; Wakabayashi, Tomonari; Matsuo, Tsukasa (2017-05-15). "Cleavage of a P=P Double Bond Mediated by N-Heterocyclic Carbenes". Angewandte Chemie International Edition. 56 (21): 5765–5769. doi:10.1002/anie.201701201. ISSN 1521-3773.
- ↑ Flynn, Kathy M.; Olmstead, Marilyn M.; Power, Philip P. (1983-04-01). "Simple one-step route to a transition-metal complex containing a phosphorus-phosphorus double bond. Synthesis and x-ray crystal structure of {trans-bis[bis(trimethylsilyl)methyl]diphosphene}bis[tetracarbonyliron(0)]". Journal of the American Chemical Society. 105 (7): 2085–2086. doi:10.1021/ja00345a080. ISSN 0002-7863.
- ↑ Cowley, A. H.; Kilduff, J. E.; Lasch, J. G.; Norman, N. C.; Pakulski, M.; Ando, F.; Wright, T. C. (1983-12-01). "Reactivity of diphosphenes and phosphaarsenes toward metal carbonyls". Journal of the American Chemical Society. 105 (26): 7751–7752. doi:10.1021/ja00364a051. ISSN 0002-7863.
- ↑ Chatt, Joseph; Hitchcock, Petter B.; Pidcock, Alan; Warrens, Christopher P.; Dixon, Keith R. (1982-01-01). "Synthesis and31P n.m.r. spectroscopy of platinum and palladium complexes containing side-bonded diphenyldiphosphene. The X-ray crystal and molecular structure of [Pd(PhPpph){bis(diphenyl-phosphino)ethane}]". J. Chem. Soc., Chem. Commun. 0 (16): 932–933. doi:10.1039/c39820000932. ISSN 0022-4936.
- ↑ Yoshifuji, Masaaki; Inamoto, Naoki. "Reaction of a diaryldiphosphene with hexacarbonylchromium(0): formation of (arene)tricarbonylchromium(0) complexes". Tetrahedron Letters. 24 (44): 4855–4858. doi:10.1016/s0040-4039(00)94025-5.
- ↑ Caminade, A. M.; Khatib, F. E.; Ades, C.; Verrier, M.; Paillous, N.; Koenig, M. (1987-04-14). "ChemInform Abstract: Oxidation and Isomerization of Diphosphene". ChemInform. 18 (15): no–no. doi:10.1002/chin.198715272. ISSN 1522-2667.
- ↑ Caminade, Anne-Marie; Couret, Claude; Escudie, Jean; Koenig, Max (1984-01-01). "Ozonolysis of bis[tris(trimethylsilyl)methyl]diphosphene". Journal of the Chemical Society, Chemical Communications. 0 (24). doi:10.1039/c39840001622. ISSN 0022-4936.
- ↑ Yoshifuji, Masaaki; Shibayama, Katsuhiro; Inamoto, Naoki; Watanabe, Tokuko (1983-04-05). "REDUCTION OF DIPHOSPHENE: FORMATION OF dl- AND meso-DIPHOSPHANES". Chemistry Letters. 12 (4): 585–588. doi:10.1246/cl.1983.585. ISSN 0366-7022.