Classical nucleation theory
Nucleation is the first step in the formation of either a new thermodynamic phase or a new structure with lower free energy via self-assembly or self-organisation. Nucleation is typically defined to be the process that determines how long we have to wait before the new phase or self-organised structure appears. Classical nucleation theory (CNT) is the most common theoretical model used to understand why nucleation may take hours, years, or never happen at all.[1][2][3]
Outline of classical nucleation theory
This is the standard simple theory for nucleation of a new thermodynamic phase, such as a liquid or a crystal. It should be borne in mind that it is approximate. The basic CNT nucleation of a new phase provides an approximate but physically reasonable prediction for the rate at which nuclei of a new phase form, via nucleation on a set of identical nucleation sites. This rate, R is the number of, for example, water droplets nucleating in a uniform volume of air supersaturated with water vapour, per unit time. So if a 100 droplets nucleate in a volume of 0.1m3 in 1s, then the rate R=1000/s. The description here follows modern standard CNT.[3] The prediction for the rate R is

where
- is the free energy cost of the nucleus at the top of the nucleation barrier, and kBT is the thermal energy with T the absolute temperature and kB is the Boltzmann constant.
- is the number of nucleation sites.
- is the rate at which molecules attach to the nucleus causing it to grow.
- is what is called the Zeldovich factor Z. Essentially the Zeldovich factor is the probability that a nucleus at the top of the barrier will go on to form the new phase, not dissolve.




This expression for the rate can be thought of as a product of two factors: The first, , is the number of nucleation sites multiplied by the probability that a nucleus of critical size has grown around it. It can be interpreted as the average, instantaneous number of nuclei at the top of the nucleation barrier. Free energies and probabilities are closely related in general, by definition.[4] The probability of a nucleus forming at a site is proportional to . So if is large and positive the probability of forming a nucleus is very low and nucleation will be slow. Then the average number will be much less than one, i.e., it is likely that at any given time none of the sites has a nucleus.

![\exp[-\Delta G^*/kT]](../I/m/2aa851977e60d3d4673586f9d4f6d00a716934a7.svg)
The second factor in the expression for the rate is the dynamic part, . Here, expresses the rate of incoming matter and is the probability that a nucleus of critical size (at the maximum of the energy barrier) will continue to grow and not dissolve. The Zeldovich factor is derived by assuming that the nuclei near the top of the barrier are effectively diffusing along the radial axis. By statistical fluctuations, a nucleus at the top of the barrier can grow diffusively into a larger nucleus that will grow into a new phase, or it can lose molecules and shrink back to nothing. The probability that a given nucleus goes forward is .

To see how this works in practice we can look at an example. Sanz and coworkers[5] have used computer simulation to estimate all the quantities in the above equation, for the nucleation of ice in liquid water. They did this for a simple but approximate model of water called TIP4P/2005. At a supercooling of 19.5 °C, i.e., 19.5 °C below the freezing point of water in their model, they estimate a free energy barrier to nucleation of ice of . They also estimate a rate of addition of water molecules to an ice nucleus near the top of the barrier of j = 1011/s and a Zeldovich factor Z = 10−3 (note that this factor is dimensionless because it is basically a probability). The number of water molecules in 1 m3 of water is approximately 1028. Putting all these numbers into the formula we get a nucleation rate of approximately 10−83/s. This means that on average we would have to wait 1083s (1076 years) to see a single ice nucleus forming in 1 m3 of water at -20 °C!

This is a rate of homogeneous nucleation estimated for a model of water, not real water—in experiments we cannot grow nuclei of water and so cannot directly determine the values of the barrier ΔG*, or the dynamic parameters such as j, for real water. However, it may be that indeed the homogeneous nucleation of ice at temperatures near -20 °C and above is extremely slow and so that whenever we see water freezing temperatures of -20 °C and above this is due to heterogeneous nucleation, i.e., the ice nucleates in contact with a surface.
Homogeneous nucleation
Homogeneous nucleation is much rarer than heterogeneous nucleation.[1][6] However, homogeneous nucleation is simpler and easier to understand than heterogeneous nucleation, so the easiest way to understand heterogeneous nucleation is to start with homogeneous nucleation. So we will outline the CNT calculation for the homogeneous nucleation barrier .
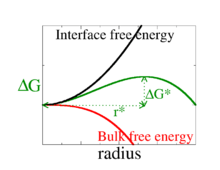

To understand if nucleation is fast or slow, needs to be calculated. The classical theory[7] assumes that even for a microscopic nucleus of the new phase, we can write the free energy of a droplet as the sum of a bulk term that is proportional to the volume of the nucleus, and a surface term, that is proportional to its surface area



The first term is the volume term, and as we are assuming that the nucleus is spherical, this is the volume of a sphere of radius . is the difference in free energy per unit volume between the thermodynamic phase nucleation is occurring in, and the phase that is nucleating. For example, if water is nucleating in supersaturated air, then is the free energy per unit volume of the supersaturated air minus that of water at the same pressure. As nucleation only occurs when the air is supersaturated, is always negative. The second term comes from the interface at surface of the nucleus, which is why it is proportional to the surface area of a sphere. is the surface tension of the interface between the nucleus and its surroundings, which is always positive.



For small the second surface term dominates and . The free energy is the sum of an and terms. Now the terms varies more rapidly with than the term, so as small the term dominates and the free energy is positive while for large , the term dominates and the free energy is negative. This shown in the figure to the right. Thus at some intermediate value of , the free energy goes through a maximum, and so the probability of formation of a nucleus goes through a minimum. There is a least-probable nucleus occurs, i.e., the one with the highest value of where




Addition of new molecules to nuclei larger than this critical radius decreases the free energy, so these nuclei are more probable. The rate at which nucleation occurs is then limited by, i.e., determined by the probability, of forming the critical nucleus. This is just the exponential of minus the free energy of the critical nucleus , which is

This is the free energy barrier needed in the CNT expression for above.

From an experimental standpoint, this theory grants tuning of the critical radius through the dependence of on temperature. The variable , described above, can be expressed as

where is the melting point and is the enthalpy of formation for the material. Furthermore, the critical radius can be expressed as



revealing a dependence of reaction temperature. Thus as you increase the temperature near , the critical radius will increase.
Heterogeneous nucleation
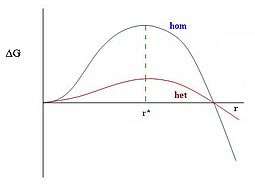
Heterogeneous nucleation is the second of the two nucleations as discussed in the classical nucleation theory. Unlike homogeneous nucleation, heterogeneous nucleation occurs when the nucleus is on the surface. As previously stated, heterogeneous nucleation is more common than homogeneous nucleation. This is due to the different factors within a single system. These factors may include a lower level of supersaturation or being more kinetically favorable than homogeneous nucleation.[8] Heterogeneous nucleation is more kinetically favorable than homogeneous nucleation because the nucleation barrier for the heterogeneous nucleation, ΔG*, is much lower at the surface in contrast to the nucleation barrier for homogeneous nucleation. This is because the nucleation barrier comes from the positive term in the free energy ΔG, which is the surface term. For homogeneous nucleation the nucleus is approximated by a sphere and so has a surface term containing the surface area of a sphere and so has a free energy equal to the surface area of a sphere, 4πr2, times the surface tension σ. However, because heterogeneous nucleation is the nucleation of a nucleus on a surface, it is important to take into account of more factors that go into a heterogeneous nucleation.
.svg.png)
As shown in a diagram on the lefthand side, the factors are the size of the droplet, the contact angle at which the particle is making contact on the surface, and the interactions at three different phases(the interaction between the liquid interface and the solid surface, the interaction between the solid surface and the air, and the interaction between the liquid interface and the air). A schematic on the righthand side shows that the macroscopic droplets are not complete spheres. Therefore, the area of the interface between the droplet and the surrounding fluid is less than . This geometrical factor reduces the interfacial area and so the interfacial free energy, which also reduces the nucleation barrier.[3] Note that this simple theory treats the microscopic nucleus just as if it is a macroscopic droplet.

In the schematic to the right the contact angle between the droplet surface and the surface decreases from left to right (A to C). In this schematic, the surface area of the droplet decreases as the contact angle decreases. This geometrical effect reduces the barrier which will increase the rate of nucleation as opposed to having a surface with a larger contact angle. Also, if instead of the surface being flat it curves like a fluid, then this also reduces the interfacial area and the nucleation barrier. There are expressions for this reduction for simple surface geometries.[9] In practice, this means we expect nucleation to be fastest on any imperfections in the surface such that the nucleus forms a small contact angle on its surface.
Comparison between CNT, computer simulation and experiment
The classical nucleation theory makes a number of assumptions, for example it treats a microscopic nucleus as if it is a macroscopic droplet with a well defined surface whose free energy is estimated using an equilibrium property: the interfacial tension σ. For a nucleus that may be only of order ten molecules across it is not always clear that we can treat something so small as a volume plus a surface. Also nucleation is an inherently out of thermodynamic equilibrium phenomenon so it is not always obvious that its rate can estimated using equilibrium properties.
For simple model systems, modern computers are powerful enough to calculate numerically exact nucleation rates. One such example is the nucleation of the crystal phase in the model of hard spheres. This is a simple model of some colloids consisting of perfectly hard spheres in thermal motion. The agreement of CNT with the calculated rates for this system confirms that the classical theory is a very reasonable approximate theory.[10] For the simple models CNT works quite well, however it is unclear if it describes complex (e.g. molecular) systems equally well. Jones et al. computationally explored the nucleation of small Water cluster using classical water model. It was found that CNT could describe the nucleation of clusters of 8-50 water molecules well, but failed to describe smaller clusters.[11] Corrections to CNT, obtained from higher accuracy methods such as quantum chemical calculations, can provide necessary interactions for accurate nucleation rates.[12] However, the CNT fails in describing experimental results of vapour to liquid nucleation even for model substances like Argon by several orders of magnitude.[13]
References
- 1 2 H. R. Pruppacher and J. D. Klett, Microphysics of Clouds and Precipitation, Kluwer (1997)
- ↑ P.G. Debenedetti, Metastable Liquids: Concepts and Principles, Princeton University Press (1997)
- 1 2 3 Sear, R. P. (2007). "Nucleation: theory and applications to protein solutions and colloidal suspensions". J. Phys.: Condens. Matter. 19 (3): 033101. Bibcode:2007JPCM...19c3101S. doi:10.1088/0953-8984/19/3/033101.
- ↑ Frenkell, Daan; Smit, Berent (2001). Understanding Molecular Simulation, Second Edition: From Algorithms to Applications. p. Academic Press. ISBN 0122673514.
- ↑ Sanz, Eduardo; Vega, Carlos; Espinosa, J. R.; Cabellero-Bernal, R.; Abascal, J. L. F.; Valeriani, Chantal (2013). "Homogeneous Ice Nucleation at Moderate Supercooling from Molecular Simulation". Journal of the American Chemical Society. 135 (40): 15008–15017. arXiv:1312.0822. doi:10.1021/ja4028814. PMID 24010583.
- ↑ Sear, Richard P. (2014). "Quantitative Studies of Crystal Nucleation at Constant Supersaturation: Experimental Data and Models" (PDF). CrystEngComm. 16 (29): 6506–6522. doi:10.1039/C4CE00344F.
- ↑ F. F. Abraham (1974) Homogeneous nucleation theory (Academic Press, NY).
- ↑ Liu, X. Y. (31 May 2000). "Heterogeneous nucleation or homogeneous nucleation?". The Journal of Chemical Physics. 112 (22): 9949–9955. Bibcode:2000JChPh.112.9949L. doi:10.1063/1.481644. ISSN 0021-9606. Retrieved 16 November 2017.
- ↑ Sholl, C. A.; N. H. Fletcher (1970). "Decoration criteria for surface steps". Acta Metall. 18 (10): 1083–1086. doi:10.1016/0001-6160(70)90006-4.
- ↑ Auer, S.; D. Frenkel (2004). "Numerical prediction of absolute crystallization rates in hard-sphere colloids". J. Chem. Phys. 120 (6): 3015. Bibcode:2004JChPh.120.3015A. doi:10.1063/1.1638740.
- ↑ Merikanto, Joonas; Zapadinsky, Evgeni; Lauri, Antti; Vehkamäki, Hanna (4 April 2007). "Origin of the Failure of Classical Nucleation Theory: Incorrect Description of the Smallest Clusters". Physical Review Letters. 98 (14). Bibcode:2007PhRvL..98n5702M. doi:10.1103/PhysRevLett.98.145702.
- ↑ Temelso, Berhane; Morrell, Thomas E.; Shields, Robert M.; Allodi, Marco A.; Wood, Elena K.; Kirschner, Karl N.; Castonguay, Thomas C.; Archer, Kaye A.; Shields, George C. (22 February 2012). "Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications". The Journal of Physical Chemistry A. 116 (9): 2209–2224. Bibcode:2012JPCA..116.2209T. doi:10.1021/jp2119026.
- ↑ A. Fladerer, R. Strey: „Homogeneous nucleation and droplet growth in supersaturated argon vapor: The cryogenic nucleation pulse chamber.“ in: The Journal of Chemical Physics 124(16), 164710 (2006). (Online)