Bond hardening
Bond hardening is a process of creating a new chemical bond by strong laser fields—an effect opposite to bond softening. However, it is not opposite in the sense that the bond becomes stronger, but in the sense that the molecule enters a state that is diametrically opposite to the bond-softened state. Such states require laser pulses of high intensity, in the range of 1013–1015 W/cm2, and they disappear once the pulse is gone.
Theory
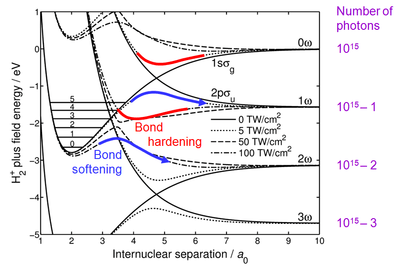
Bond hardening and bond softening share the same theoretical basis, which is described under the latter entry. Briefly, the ground and the first excited energy curves of the H2+ ion are dressed in photons. The laser field perturbs the curves and turns their crossings into anticrossings. Bond softening occurs on the lower branches of the anticrossings and bond hardening happens if the molecule is excited to the upper branches – see Fig. 1.
To trap the molecule in the bond-hardened state, the anticrossing gap cannot be too small or too large. If it is too small, the system can undergo a diabatic transition to the lower branch of the anticrossing and dissociate via bond softening. If the gap is too large, the upper branch becomes shallow or even repulsive, and the system can also dissociate. This means that bound bond-hardened states can exist only in relatively narrow range of laser intensities, which makes them difficult to observe.
Experimental search for bond hardening
When the existence of bond softening was experimentally verified in 1990,[1] the attention turned to bond hardening. Rather noisy photoelectron spectra reported in the early 1990s implied bond hardening occurring at the 1-photon[2] and 3-photon[3] anticrossings. These reports were received with great interest because bond hardening could explain apparent stabilization of the molecular bond in strong laser fields[4] accompanied by a collective ejection of several electrons.[5] However, instead of more convincing evidence, new negative results relegated bond hardening to a remote theoretical possibility.[6] Only at the end of the decade, the reality of bond hardening was established in an experiment[7] where the laser pulse duration was varied by chirping.
Conclusive evidence
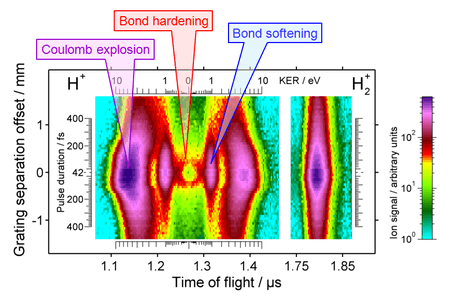
The results of the chirp experiment are shown in Fig. 2 in the form of a map. The central "crater" of the map is a signature of bond hardening. To appreciate the uniqueness of this signature requires explaining other features on the map.
The horizontal axis of the map gives the time-of-flight (TOF) of ions produced in ionization and fragmentation of molecular hydrogen exposed to intense laser pulses. The left panel reveals several proton peaks; the right panel shows relatively uninteresting, single peak of molecular hydrogen ion.
The vertical axis gives grating position of the compressor in a chirped pulse amplifier of the Ti:Sapphire laser used in the experiment. The grating position controls the pulse duration, which is shortest (42 fs) for the zero position and increases in both directions. While the stretched pulses are also chirped, it is not the chirp but the pulse duration that matters in this experiment, as corroborated by the symmetry of the map in respect to the zero position line. The pulse energy is kept constant, therefore the shortest pulses are also most intense producing most ions at the zero position.
Kinetic energy variation
The proton TOF spectra allow one to measure the kinetic energy release (KER) in the dissociation process. Protons ejected towards the detector have shorter TOFs than protons ejected away from the detector because the latter have to be turned back by an external electric field applied to the interaction region. This forward-backward symmetry is reflected in the symmetry of the proton map in respect to zero KER (1.27 µs TOF).
The most energetic protons come from the Coulomb explosion of the molecule, where laser field completely strips H2 from electrons and the two bare protons repel each other with strong Coulombic force, unimpeded by any chemical bond. The stripping process it not instantaneous but occurs in a stepwise fashion,[8] on the rising edge of the laser pulse. The shorter the laser pulse, the quicker the stripping process and there is less time for the molecule to dissociate before the Coulomb force attains its full strength. Therefore, the KER is highest for the shortest pulses, as demonstrated by the outer curving "lobes" in Fig. 2.
The second pair of proton peaks (1 eV KER) comes from bond softening of the H2+ ion, which dissociates into a proton and a neutral hydrogen atom (undetected). The dissociation starts at the 3-photon gap and proceeds to the 2ω limit (the lower blue arrow in Fig. 1). Since both the initial and the final energies of this process are fixed by the 1.55 eV photon energy, the KER is also constant producing the two vertical lines in Fig. 2.
The lowest energy protons are produced by the bond hardening process, which also starts at the 3-photon gap but proceeds to the 1ω limit (the lower red trough in Fig. 1). Since the initial and the final energies are also fixed here, the KER should also be constant but clearly it is not, as the round shape of the central "crater" demonstrates it in Fig. 2. To explain this variation, the dynamics of the H2+ states needs to be considered.
Dynamics of bond hardening
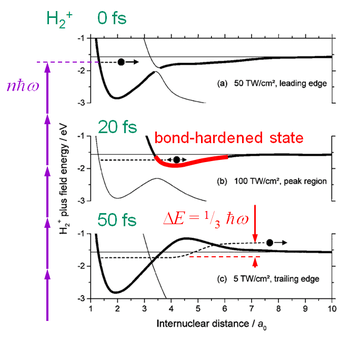
The H2+ ion is created on the leading edge of the laser pulse in the multiphoton ionization process. Since the equilibrium internuclear separation for the neutral molecule is smaller than for the ionized one, the ionic nuclear wave packet finds itself on the repulsive side of the ground state potential well and starts to cross it (see Fig. 3a).
In a few femtoseconds it takes the wave packet to cross the potential well, the laser intensity is still modest and the 3-photon gap is small allowing the wave packet to cross it diabatically. At large internuclear separations, the gentle slope of the potential well slowly turns the wave packet back, so when the packet returns to the 3-photon gap, the laser intensity is significantly higher and the gap is wide open trapping the wave packet in a bond-hardened state, which lasts throughout the highest intensities (Fig. 3b).
When the laser intensity falls, the bond-hardened energy curve returns to the original shape, flexing up, lifting the wave packet and releasing about a half of it to the 1ω limit (Fig. 3c). The faster intensity falls, the higher the wave packet is lifted and more energy it gains, which explains why the KER of the "crater" in Fig. 1 is highest at the shortest laser pulse. This energy gain, however, is not induced by the rising edge of the laser pulse as one would naively expect, but by the falling edge.
A fraction of a photon?
Note that the maximum energy gain of the nuclear wave packet is about 1⁄3ħω and continuously decreases with the pulse duration. Does it mean we can have a fraction of a photon? There are two valid answers to this puzzling proposition.
Breakdown of the photon model
One can say that the photon is not a particle but as a mere quantum of energy that is usually exchanged in integer multiples of ħω, but not always, as it is the case in the above experiment. From this point of view, photons are quasiparticles, akin to phonons and plasmons, in a sense less "real" than electrons and protons. Before dismissing this view as unscientific, its worth recalling the words of Willis Lamb, who won a Nobel prize in the area of quantum electrodynamics:
There is no such thing as a photon. Only a comedy of errors and historical accidents led to its popularity among physicists and optical scientists.[9]
Dynamic Raman effect
Alternatively, one can save the photon concept by recalling that the laser field is very strong and the pulse is very short. Indeed, the electric field in the laser pulse is so strong that during the process depicted in Fig. 3 about a hundred of photon absorptions and stimulated emissions can take place. And since the pulse is short, it has sufficiently wide bandwidth to accommodate absorption of photons that are more energetic than the re-emitted ones, giving the net result of a fraction of ħω. Effectively, we have a kind of dynamic Raman effect.
Zero-photon dissociation
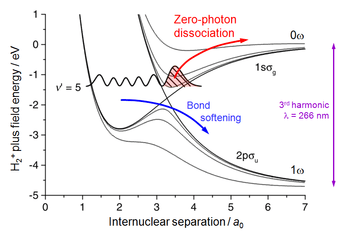
Even more striking challenge to the photon concept comes from the zero-photon dissociation process (ZPD), where nominally no photons are absorbed but some energy is still extracted from the laser field. To demonstrate this process, molecular hydrogen was exposed to 250 fs pulses of the 3rd harmonic of a Ti:Sapphire laser.[10] Since the photon energy was 3 times higher, the spacing of the energy curves shown in Fig. 1 was 3 times larger, replacing the 3-photon crossing with a 1-photon one, as shown in Fig. 4. As before, the laser field changed the crossing to anticrossing, bond softening was induced on its lower branch and bond hardening trapped a part of the vibrational wave packet on the upper branch. In increasing laser intensity the anticrossing gap was getting wider, lifting the wave packet to the 0ω limit and dissociating the molecule with very small KER.
The experimental signature[10] of the ZPD was a proton peak at zero KER. Moreover, the probability of a proton being promoted to this peak was found to be independent of the laser intensity, which confirms that it is induced by a zero-photon process because the probability of multiphoton processes is proportional to the intensity, I, raised to the number of photons absorbed, giving I0 = const.
See also
- Conical intersections of energy surfaces in polyatomic molecules share many similarities with the simpler mechanism of bond hardening and bond softening in diatomic molecules.
References
- ↑ P.H. Bucksbaum, A. Zavriyev, H.G. Muller and D.W. Schumacher "Softening of the H2+ molecular bond in intense laser fields" Phys. Rev. Lett. 64 1883 (1990)
- ↑ S.W. Allendorf and A. Szöke "High-intensity multiphoton ionization of H2" Phys. Rev. A 44 518 (1991)
- ↑ A. Zavriyev, P.H. Bucksbaum, J. Squier and F. Saline "Light-Induced Vibrational Structure in H2+ and D2+ in Intense Laser Fields" Phys. Rev. Lett. 70 1077 (1993)
- ↑ K. Codling and L. J. Frasinski "Topical review: Dissociative ionization of small molecules in intense laser fields" J. Phys. B 26 783 (1993); M. Schmidt, D. Normand and C. Cornaggia "Laser-induced trapping of chlorine molecules with pico- and femtosecond pulses" Phys. Rev. A 50 5037 (1994)
- ↑ L.J. Frasinski, K. Codling, P. Hatherly, J. Barr, I.N. Ross and W.T. Toner "Femtosecond Dynamics of Multielectron Dissociative Ionization by Use of a Picosecond Laser" Physical Review Letters 58 2424–2427 (1987), open access
- ↑ T.D.G. Walsh, F.A. Ilkov and S.L. Chin "The dynamical behaviour of H2 and D2 in a strong, femtosecond, titanium:sapphire laser field" J. Phys. B 30 2167 (1997); G.N. Gibson, M. Li, C. Guo and J. Neira Phys. Rev. Lett. 79 2022 (1997)
- ↑ L.J. Frasinski, J.H. Posthumus, J. Plumridge, K. Codling, P.F. Taday and A.J. Langley "Manipulation of bond hardening in H2+ by chirping of intense femtosecond laser pulses" Phys. Rev. Lett. 83 3625–3628 (1999), open access
- ↑ K. Codling, L.J. Frasinski, P. Hatherly and J.R.M. Barr "On the major mode of multiphoton multiple ionisation" J. Phys. B 20 L525–L531 (1987)
- ↑ W.E. Lamb, Jr. "Anti-photon" Appl. Phys. B 60 77–84 (1995), http://www-3.unipv.it/fis/tamq/Anti-photon.pdf
- 1 2 J.H. Posthumus, J. Plumridge, L.J. Frasinski, K. Codling, E.J. Divall, A.J. Langley and P.F. Taday "Slow protons as a signature of zero-photon dissociation of H2+ in intense laser fields" J. Phys. B 33 L563–L570 (2000)