Calcium carbonate
Calcium carbonate is a chemical compound with the formula CaCO3. It is a common substance found in rocks as the minerals calcite and aragonite (most notably as limestone, which is a type of sedimentary rock consisting mainly of calcite) and is the main component of pearls and the shells of marine organisms, snails, and eggs. Calcium carbonate is the active ingredient in agricultural lime and is created when calcium ions in hard water react with carbonate ions to create limescale. It is medicinally used as a calcium supplement or as an antacid, but excessive consumption can be hazardous and cause poor digestion.
| |||
 | |||
| Names | |||
|---|---|---|---|
| IUPAC name
Calcium carbonate | |||
| Other names | |||
| Identifiers | |||
3D model (JSmol) |
|||
| ChEBI | |||
| ChEMBL | |||
| ChemSpider | |||
| ECHA InfoCard | 100.006.765 | ||
| EC Number |
| ||
| E number | E170 (colours) | ||
| KEGG | |||
PubChem CID |
|||
| RTECS number |
| ||
| UNII | |||
CompTox Dashboard (EPA) |
|||
InChI
| |||
| |||
| Properties | |||
| CaCO3 | |||
| Molar mass | 100.0869 g/mol | ||
| Appearance | Fine white powder; chalky taste | ||
| Odor | odorless | ||
| Density | 2.711 g/cm3 (calcite) 2.83 g/cm3 (aragonite) | ||
| Melting point | 1,339 °C (2,442 °F; 1,612 K) (calcite) 825 °C (1,517 °F; 1,098 K) (aragonite)[1][2] | ||
| Boiling point | decomposes | ||
| 0.013 g/L (25 °C)[3][4] | |||
Solubility product (Ksp) |
3.3×10−9[5] | ||
| Solubility in dilute acids | soluble | ||
| Acidity (pKa) | 9.0 | ||
| −3.82×10−5 cm3/mol | |||
Refractive index (nD) |
1.59 | ||
| Structure | |||
| Trigonal | |||
Space group |
32/m | ||
| Thermochemistry | |||
Std molar entropy (S |
93 J·mol−1·K−1[6] | ||
Std enthalpy of formation (ΔfH⦵298) |
−1207 kJ·mol−1[6] | ||
| Pharmacology | |||
| A02AC01 (WHO) A12AA04 (WHO) | |||
| Hazards | |||
| Safety data sheet | ICSC 1193 | ||
| NFPA 704 (fire diamond) | 
0
0
0 | ||
| Lethal dose or concentration (LD, LC): | |||
LD50 (median dose) |
6450 mg/kg (oral, rat) | ||
| NIOSH (US health exposure limits): | |||
PEL (Permissible) |
TWA 15 mg/m3 (total) TWA 5 mg/m3 (resp)[7] | ||
| Related compounds | |||
Other anions |
Calcium bicarbonate | ||
Other cations |
Magnesium carbonate Strontium carbonate Barium carbonate | ||
Related compounds |
Calcium sulfate | ||
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |||
| Infobox references | |||


Chemistry
Calcium carbonate shares the typical properties of other carbonates. Notably it
- reacts with acids, releasing carbon dioxide (technically speaking, carbonic acid, but that disintegrates quickly to CO2 and H2O):
- CaCO3(s) + 2 H+(aq) → Ca2+(aq) + CO2(g) + H2O(l)
- releases carbon dioxide upon heating, called a thermal decomposition reaction, or calcination (to above 840 °C in the case of CaCO3), to form calcium oxide, commonly called quicklime, with reaction enthalpy 178 kJ/mol:
- CaCO3(s) → CaO(s) + CO2(g)
Calcium carbonate will react with water that is saturated with carbon dioxide to form the soluble calcium bicarbonate.
- CaCO3(s) + CO2(g) + H2O(l) → Ca(HCO3)2(aq)
This reaction is important in the erosion of carbonate rock, forming caverns, and leads to hard water in many regions.
An unusual form of calcium carbonate is the hexahydrate, ikaite, CaCO3·6H2O. Ikaite is stable only below 8 °C.
Preparation
The vast majority of calcium carbonate used in industry is extracted by mining or quarrying. Pure calcium carbonate (such as for food or pharmaceutical use), can be produced from a pure quarried source (usually marble).
Alternatively, calcium carbonate is prepared from calcium oxide. Water is added to give calcium hydroxide then carbon dioxide is passed through this solution to precipitate the desired calcium carbonate, referred to in the industry as precipitated calcium carbonate (PCC):[8]
- CaO + H2O → Ca(OH)2
- Ca(OH)2 + CO2 → CaCO3↓ + H2O
Structure
The thermodynamically stable form of CaCO3 under normal conditions is hexagonal β-CaCO3 (the mineral calcite).[9] Other forms can be prepared, the denser (2.83 g/cm3) orthorhombic λ-CaCO3 (the mineral aragonite) and hexagonal μ-CaCO3, occurring as the mineral vaterite.[9] The aragonite form can be prepared by precipitation at temperatures above 85 °C, the vaterite form can be prepared by precipitation at 60 °C.[9] Calcite contains calcium atoms coordinated by six oxygen atoms, in aragonite they are coordinated by nine oxygen atoms.[9] The vaterite structure is not fully understood.[10] Magnesium carbonate (MgCO3) has the calcite structure, whereas strontium carbonate and barium carbonate (SrCO3 and BaCO3) adopt the aragonite structure, reflecting their larger ionic radii.[9]
Occurrence

Geological sources
Calcite, aragonite and vaterite are pure calcium carbonate minerals. Industrially important source rocks which are predominantly calcium carbonate include limestone, chalk, marble and travertine.
Biological sources

Eggshells, snail shells and most seashells are predominantly calcium carbonate and can be used as industrial sources of that chemical.[12] Oyster shells have enjoyed recent recognition as a source of dietary calcium, but are also a practical industrial source.[13][14] Dark green vegetables such as broccoli and kale contain dietarily significant amounts of calcium carbonate, however, they are not practical as an industrial source.[15]
Geology

Carbonate is found frequently in geologic settings and constitutes an enormous carbon reservoir. Calcium carbonate occurs as aragonite, calcite and dolomite as significant constituents of the calcium cycle. The carbonate minerals form the rock types: limestone, chalk, marble, travertine, tufa, and others.
In warm, clear tropical waters corals are more abundant than towards the poles where the waters are cold. Calcium carbonate contributors, including plankton (such as coccoliths and planktic foraminifera), coralline algae, sponges, brachiopods, echinoderms, bryozoa and mollusks, are typically found in shallow water environments where sunlight and filterable food are more abundant. Cold-water carbonates do exist at higher latitudes but have a very slow growth rate. The calcification processes are changed by ocean acidification.
Where the oceanic crust is subducted under a continental plate sediments will be carried down to warmer zones in the asthenosphere and lithosphere. Under these conditions calcium carbonate decomposes to produce carbon dioxide which, along with other gases, give rise to explosive volcanic eruptions.
Carbonate compensation depth
The carbonate compensation depth (CCD) is the point in the ocean where the rate of precipitation of calcium carbonate is balanced by the rate of dissolution due to the conditions present. Deep in the ocean, the temperature drops and pressure increases. Calcium carbonate is unusual in that its solubility increases with decreasing temperature.[18] Increasing pressure also increases the solubility of calcium carbonate. The carbonate compensation depth can range from 4,000 to 6,000 meters below sea level.
Role in taphonomy
Calcium carbonate can preserve fossils through permineralization. Most of the vertebrate fossils of the Two Medicine Formation—a geologic formation known for its duck-billed dinosaur eggs—are preserved by CaCO3 permineralization.[19] This type of preservation conserves high levels of detail, even down to the microscopic level. However, it also leaves specimens vulnerable to weathering when exposed to the surface.[19]
Trilobite populations were once thought to have composed the majority of aquatic life during the Cambrian, due to the fact that their calcium carbonate-rich shells were more easily preserved than those of other species,[20] which had purely chitinous shells.
Uses
Industrial applications
The main use of calcium carbonate is in the construction industry, either as a building material, or limestone aggregate for road building, as an ingredient of cement, or as the starting material for the preparation of builders' lime by burning in a kiln. However, because of weathering mainly caused by acid rain,[21] calcium carbonate (in limestone form) is no longer used for building purposes on its own, but only as a raw primary substance for building materials.
Calcium carbonate is also used in the purification of iron from iron ore in a blast furnace. The carbonate is calcined in situ to give calcium oxide, which forms a slag with various impurities present, and separates from the purified iron.[22]
In the oil industry, calcium carbonate is added to drilling fluids as a formation-bridging and filtercake-sealing agent; it is also a weighting material which increases the density of drilling fluids to control the downhole pressure. Calcium carbonate is added to swimming pools, as a pH corrector for maintaining alkalinity and offsetting the acidic properties of the disinfectant agent.[23]
It is also used as a raw material in the refining of sugar from sugar beet; it is calcined in a kiln with anthracite to produce calcium oxide and carbon dioxide. This burnt lime is then slaked in fresh water to produce a calcium hydroxide suspension for the precipitation of impurities in raw juice during carbonatation.[24]
Calcium carbonate in the form of chalk has traditionally been a major component of blackboard chalk. However, modern manufactured chalk is mostly gypsum, hydrated calcium sulfate CaSO4·2H2O. Calcium carbonate is a main source for growing Seacrete. Precipitated calcium carbonate (PCC), pre-dispersed in slurry form, is a common filler material for latex gloves with the aim of achieving maximum saving in material and production costs.[25]
Fine ground calcium carbonate (GCC) is an essential ingredient in the microporous film used in diapers and some building films, as the pores are nucleated around the calcium carbonate particles during the manufacture of the film by biaxial stretching. GCC and PCC are used as a filler in paper because they are cheaper than wood fiber. In terms of market volume, GCC are the most important types of fillers currently used.[26] Printing and writing paper can contain 10–20% calcium carbonate. In North America, calcium carbonate has begun to replace kaolin in the production of glossy paper. Europe has been practicing this as alkaline papermaking or acid-free papermaking for some decades. PCC used for paper filling and paper coatings is precipitated and prepared in a variety of shapes and sizes having characteristic narrow particle size distributions and equivalent spherical diameters of 0.4 to 3 micrometers.
Calcium carbonate is widely used as an extender in paints,[27] in particular matte emulsion paint where typically 30% by weight of the paint is either chalk or marble. It is also a popular filler in plastics.[27] Some typical examples include around 15 to 20% loading of chalk in unplasticized polyvinyl chloride (uPVC) drainpipes, 5% to 15% loading of stearate-coated chalk or marble in uPVC window profile. PVC cables can use calcium carbonate at loadings of up to 70 phr (parts per hundred parts of resin) to improve mechanical properties (tensile strength and elongation) and electrical properties (volume resistivity). Polypropylene compounds are often filled with calcium carbonate to increase rigidity, a requirement that becomes important at high usage temperatures.[28] Here the percentage is often 20–40%. It also routinely used as a filler in thermosetting resins (sheet and bulk molding compounds)[28] and has also been mixed with ABS, and other ingredients, to form some types of compression molded "clay" poker chips.[29] Precipitated calcium carbonate, made by dropping calcium oxide into water, is used by itself or with additives as a white paint, known as whitewashing.[30][31]
Calcium carbonate is added to a wide range of trade and do it yourself adhesives, sealants, and decorating fillers.[27] Ceramic tile adhesives typically contain 70% to 80% limestone. Decorating crack fillers contain similar levels of marble or dolomite. It is also mixed with putty in setting stained glass windows, and as a resist to prevent glass from sticking to kiln shelves when firing glazes and paints at high temperature.
In ceramic glaze applications, calcium carbonate is known as whiting,[27] and is a common ingredient for many glazes in its white powdered form. When a glaze containing this material is fired in a kiln, the whiting acts as a flux material in the glaze. Ground calcium carbonate is an abrasive (both as scouring powder and as an ingredient of household scouring creams), in particular in its calcite form, which has the relatively low hardness level of 3 on the Mohs scale, and will therefore not scratch glass and most other ceramics, enamel, bronze, iron, and steel, and have a moderate effect on softer metals like aluminium and copper. A paste made from calcium carbonate and deionized water can be used to clean tarnish on silver.[32]
Health and dietary applications

Calcium carbonate is widely used medicinally as an inexpensive dietary calcium supplement for gastric antacid[33] (such as Tums). It may be used as a phosphate binder for the treatment of hyperphosphatemia (primarily in patients with chronic kidney failure). It is used in the pharmaceutical industry as an inert filler for tablets and other pharmaceuticals.[34]
Calcium carbonate is used in the production of calcium oxide as well as toothpaste and has seen a resurgence as a food preservative and color retainer, when used in or with products such as organic apples.[35]
Calcium carbonate is used therapeutically as phosphate binder in patients on maintenance haemodialysis. It is the most common form of phosphate binder prescribed, particularly in non-dialysis chronic kidney disease. Calcium carbonate is the most commonly used phosphate binder, but clinicians are increasingly prescribing the more expensive, non-calcium-based phosphate binders, particularly sevelamer.
Excess calcium from supplements, fortified food, and high-calcium diets can cause milk-alkali syndrome, which has serious toxicity and can be fatal. In 1915, Bertram Sippy introduced the "Sippy regimen" of hourly ingestion of milk and cream, and the gradual addition of eggs and cooked cereal, for 10 days, combined with alkaline powders, which provided symptomatic relief for peptic ulcer disease. Over the next several decades, the Sippy regimen resulted in kidney failure, alkalosis, and hypercalcaemia, mostly in men with peptic ulcer disease. These adverse effects were reversed when the regimen stopped, but it was fatal in some patients with protracted vomiting. Milk-alkali syndrome declined in men after effective treatments for peptic ulcer disease arose. Since the 1990s it has been most frequently reported in women taking calcium supplements above the recommended range of 1.2 to 1.5 grams daily, for prevention and treatment of osteoporosis,[36][37] and is exacerbated by dehydration. Calcium has been added to over-the-counter products, which contributes to inadvertent excessive intake. Excessive calcium intake can lead to hypercalcemia, complications of which include vomiting, abdominal pain and altered mental status.[38]
As a food additive it is designated E170,[39] and it has an INS number of 170. Used as an acidity regulator, anticaking agent, stabilizer or color it is approved for usage in the EU,[40] USA[41] and Australia and New Zealand.[42] It is used in some soy milk and almond milk products as a source of dietary calcium; one study suggests that calcium carbonate might be as bioavailable as the calcium in cow's milk.[43] Calcium carbonate is also used as a firming agent in many canned and bottled vegetable products.
Agricultural use
Agricultural lime, powdered chalk or limestone, is used as a cheap method for neutralising acidic soil, making it suitable for planting.[44]
Household use
Calcium carbonate is a key ingredient in many household cleaning powders like Comet and is used as a scrubbing agent.
Environmental applications
In 1989, a researcher, Ken Simmons, introduced CaCO3 into the Whetstone Brook in Massachusetts.[45] His hope was that the calcium carbonate would counter the acid in the stream from acid rain and save the trout that had ceased to spawn. Although his experiment was a success, it did increase the amount of aluminium ions in the area of the brook that was not treated with the limestone. This shows that CaCO3 can be added to neutralize the effects of acid rain in river ecosystems. Currently calcium carbonate is used to neutralize acidic conditions in both soil and water.[46][47][48] Since the 1970s, such liming has been practiced on a large scale in Sweden to mitigate acidification and several thousand lakes and streams are limed repeatedly.[49]
Calcium carbonate is also used in flue gas desulfurisation applications eliminating harmful SO2 and NO2 emissions from coal and other fossil fuels burnt in large fossil fuel power stations.[46]
Calcination equilibrium
Calcination of limestone using charcoal fires to produce quicklime has been practiced since antiquity by cultures all over the world. The temperature at which limestone yields calcium oxide is usually given as 825 °C, but stating an absolute threshold is misleading. Calcium carbonate exists in equilibrium with calcium oxide and carbon dioxide at any temperature. At each temperature there is a partial pressure of carbon dioxide that is in equilibrium with calcium carbonate. At room temperature the equilibrium overwhelmingly favors calcium carbonate, because the equilibrium CO2 pressure is only a tiny fraction of the partial CO2 pressure in air, which is about 0.035 kPa.
At temperatures above 550 °C the equilibrium CO2 pressure begins to exceed the CO2 pressure in air. So above 550 °C, calcium carbonate begins to outgas CO2 into air. However, in a charcoal fired kiln, the concentration of CO2 will be much higher than it is in air. Indeed, if all the oxygen in the kiln is consumed in the fire, then the partial pressure of CO2 in the kiln can be as high as 20 kPa.[50]
The table shows that this partial pressure is not achieved until the temperature is nearly 800 °C. For the outgassing of CO2 from calcium carbonate to happen at an economically useful rate, the equilibrium pressure must significantly exceed the ambient pressure of CO2. And for it to happen rapidly, the equilibrium pressure must exceed total atmospheric pressure of 101 kPa, which happens at 898 °C.
Equilibrium pressure of CO2 over CaCO3 (P) versus temperature (T).[51] P (kPa) 0.055 0.13 0.31 1.80 5.9 9.3 14 24 34 51 72 80 91 101 179 901 3961 T (°C) 550 587 605 680 727 748 777 800 830 852 871 881 891 898 937 1082 1241
Solubility
With varying CO2 pressure

Calcium carbonate is poorly soluble in pure water (47 mg/L at normal atmospheric CO2 partial pressure as shown below).
The equilibrium of its solution is given by the equation (with dissolved calcium carbonate on the right):
CaCO3 ⇌ Ca2+ + CO2−
3Ksp = 3.7×10−9 to 8.7×10−9 at 25 °C
where the solubility product for [Ca2+][CO2−
3] is given as anywhere from Ksp = 3.7×10−9 to Ksp = 8.7×10−9 at 25 °C, depending upon the data source.[51][52] What the equation means is that the product of molar concentration of calcium ions (moles of dissolved Ca2+ per liter of solution) with the molar concentration of dissolved CO2−
3 cannot exceed the value of Ksp. This seemingly simple solubility equation, however, must be taken along with the more complicated equilibrium of carbon dioxide with water (see carbonic acid). Some of the CO2−
3 combines with H+ in the solution according to
HCO−
3 ⇌ H+ + CO2−
3Ka2 = 5.61×10−11 at 25 °C
HCO−
3 is known as the bicarbonate ion. Calcium bicarbonate is many times more soluble in water than calcium carbonate—indeed it exists only in solution.
Some of the HCO−
3 combines with H+ in solution according to
H2CO3 ⇌ H+ + HCO−
3Ka1 = 2.5×10−4 at 25 °C
Some of the H2CO3 breaks up into water and dissolved carbon dioxide according to
H2O + CO2(aq) ⇌ H2CO3 Kh = 1.70×10−3 at 25 °C
And dissolved carbon dioxide is in equilibrium with atmospheric carbon dioxide according to
where kH = 29.76 atm/(mol/L) at 25 °C (Henry constant), PCO2 being the CO2 partial pressure.

For ambient air, PCO2 is around 3.5×10−4 atmospheres (or equivalently 35 Pa). The last equation above fixes the concentration of dissolved CO2 as a function of PCO2, independent of the concentration of dissolved CaCO3. At atmospheric partial pressure of CO2, dissolved CO2 concentration is 1.2×10−5 moles per liter. The equation before that fixes the concentration of H2CO3 as a function of CO2 concentration. For [CO2] = 1.2×10−5, it results in [H2CO3] = 2.0×10−8 moles per liter. When [H2CO3] is known, the remaining three equations together with
| PCO2 (atm) | pH | [Ca2+] (mol/L) |
|---|---|---|
| 10−12 | 12.0 | 5.19×10−3 |
| 10−10 | 11.3 | 1.12×10−3 |
| 10−8 | 10.7 | 2.55×10−4 |
| 10−6 | 9.83 | 1.20×10−4 |
| 10−4 | 8.62 | 3.16×10−4 |
| 3.5×10−4 | 8.27 | 4.70×10−4 |
| 10−3 | 7.96 | 6.62×10−4 |
| 10−2 | 7.30 | 1.42×10−3 |
| 10−1 | 6.63 | 3.05×10−3 |
| 1 | 5.96 | 6.58×10−3 |
| 10 | 5.30 | 1.42×10−2 |
H2O ⇌ H+ + OH− K = 10−14 at 25 °C
(which is true for all aqueous solutions), and the fact that the solution must be electrically neutral,
- 2 [Ca2+] + [H+] = [HCO−
3] + 2 [CO2−
3] + [OH−]
make it possible to solve simultaneously for the remaining five unknown concentrations (note that the above form of the neutrality equation is valid only if calcium carbonate has been put in contact with pure water or with a neutral pH solution; in the case where the initial water solvent pH is not neutral, the equation is modified).
The adjacent table shows the result for [Ca2+] and [H+] (in the form of pH) as a function of ambient partial pressure of CO2 (Ksp = 4.47×10−9 has been taken for the calculation).
- At atmospheric levels of ambient CO2 the table indicates that the solution will be slightly alkaline with a maximum CaCO3 solubility of 47 mg/L.
- As ambient CO2 partial pressure is reduced below atmospheric levels, the solution becomes more and more alkaline. At extremely low PCO2, dissolved CO2, bicarbonate ion, and carbonate ion largely evaporate from the solution, leaving a highly alkaline solution of calcium hydroxide, which is more soluble than CaCO3. Note that for PCO2 = 10−12 atm, the [Ca2+][OH−]2 product is still below the solubility product of Ca(OH)2 (8×10−6). For still lower CO2 pressure, Ca(OH)2 precipitation will occur before CaCO3 precipitation.
- As ambient CO2 partial pressure increases to levels above atmospheric, pH drops, and much of the carbonate ion is converted to bicarbonate ion, which results in higher solubility of Ca2+.
The effect of the latter is especially evident in day-to-day life of people who have hard water. Water in aquifers underground can be exposed to levels of CO2 much higher than atmospheric. As such water percolates through calcium carbonate rock, the CaCO3 dissolves according to the second trend. When that same water then emerges from the tap, in time it comes into equilibrium with CO2 levels in the air by outgassing its excess CO2. The calcium carbonate becomes less soluble as a result, and the excess precipitates as lime scale. This same process is responsible for the formation of stalactites and stalagmites in limestone caves.
Two hydrated phases of calcium carbonate, monohydrocalcite CaCO3·H2O and ikaite CaCO3·6H2O, may precipitate from water at ambient conditions and persist as metastable phases.
With varying pH, temperature and salinity: CaCO3 scaling in swimming pools
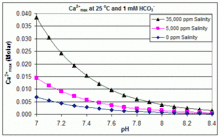
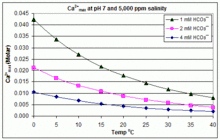
In contrast to the open equilibrium scenario above, many swimming pools are managed by addition of sodium bicarbonate (NaHCO3) to about 2 mM as a buffer, then control of pH through use of HCl, NaHSO4, Na2CO3, NaOH or chlorine formulations that are acidic or basic. In this situation, dissolved inorganic carbon (total inorganic carbon) is far from equilibrium with atmospheric CO2. Progress towards equilibrium through outgassing of CO2 is slowed by
In this situation, the dissociation constants for the much faster reactions
- H2CO3 ⇌ H+ + HCO−
3 ⇌ 2 H+ + CO2−
3
allow the prediction of concentrations of each dissolved inorganic carbon species in solution, from the added concentration of HCO−
3 (which constitutes more than 90% of Bjerrum plot species from pH 7 to pH 8 at 25 °C in fresh water).[54] Addition of HCO−
3 will increase CO2−
3 concentration at any pH. Rearranging the equations given above, we can see that [Ca2+] = Ksp/[CO2−
3], and [CO2−
3] = Ka2 [HCO−
3]/[H+]. Therefore, when HCO−
3 concentration is known, the maximum concentration of Ca2+ ions before scaling through CaCO3 precipitation can be predicted from the formula:

The solubility product for CaCO3 (Ksp) and the dissociation constants for the dissolved inorganic carbon species (including Ka2) are all substantially affected by temperature and salinity,[54] with the overall effect that [Ca2+]max increases from freshwater to saltwater, and decreases with rising temperature, pH, or added bicarbonate level, as illustrated in the accompanying graphs.
The trends are illustrative for pool management, but whether scaling occurs also depends on other factors including interactions with Mg2+, B(OH)−
4 and other ions in the pool, as well as supersaturation effects.[55][56] Scaling is commonly observed in electrolytic chlorine generators, where there is a high pH near the cathode surface and scale deposition further increases temperature. This is one reason that some pool operators prefer borate over bicarbonate as the primary pH buffer, and avoid the use of pool chemicals containing calcium.[57]
Solubility in a strong or weak acid solution
Solutions of strong (HCl), moderately strong (sulfamic) or weak (acetic, citric, sorbic, lactic, phosphoric) acids are commercially available. They are commonly used as descaling agents to remove limescale deposits. The maximum amount of CaCO3 that can be "dissolved" by one liter of an acid solution can be calculated using the above equilibrium equations.
- In the case of a strong monoacid with decreasing acid concentration [A] = [A−], we obtain (with CaCO3 molar mass = 100 g/mol):
[A] (mol/L) 1 10−1 10−2 10−3 10−4 10−5 10−6 10−7 10−10 Initial pH 0.00 1.00 2.00 3.00 4.00 5.00 6.00 6.79 7.00 Final pH 6.75 7.25 7.75 8.14 8.25 8.26 8.26 8.26 8.27 Dissolved CaCO3
(g/L of acid)50.0 5.00 0.514 0.0849 0.0504 0.0474 0.0471 0.0470 0.0470
- where the initial state is the acid solution with no Ca2+ (not taking into account possible CO2 dissolution) and the final state is the solution with saturated Ca2+. For strong acid concentrations, all species have a negligible concentration in the final state with respect to Ca2+ and A− so that the neutrality equation reduces approximately to 2[Ca2+] = [A−] yielding [Ca2+] ≈ 1/2 [A−]. When the concentration decreases, [HCO−
3] becomes non-negligible so that the preceding expression is no longer valid. For vanishing acid concentrations, one can recover the final pH and the solubility of CaCO3 in pure water.
- In the case of a weak monoacid (here we take acetic acid with pKa = 4.76) with decreasing total acid concentration [A] = [A−] + [AH], we obtain:
[A] (mol/L) 1 10−1 10−2 10−3 10−4 10−5 10−6 10−7 10−10 Initial pH 2.38 2.88 3.39 3.91 4.47 5.15 6.02 6.79 7.00 Final pH 6.75 7.25 7.75 8.14 8.25 8.26 8.26 8.26 8.27 Dissolved CaCO3
(g/L of acid)49.5 4.99 0.513 0.0848 0.0504 0.0474 0.0471 0.0470 0.0470
- For the same total acid concentration, the initial pH of the weak acid is less acid than the one of the strong acid; however, the maximum amount of CaCO3 which can be dissolved is approximately the same. This is because in the final state, the pH is larger than the pKa, so that the weak acid is almost completely dissociated, yielding in the end as many H+ ions as the strong acid to "dissolve" the calcium carbonate.
- The calculation in the case of phosphoric acid (which is the most widely used for domestic applications) is more complicated since the concentrations of the four dissociation states corresponding to this acid must be calculated together with [HCO−
3], [CO2−
3], [Ca2+], [H+] and [OH−]. The system may be reduced to a seventh degree equation for [H+] the numerical solution of which gives
[A] (mol/L) 1 10−1 10−2 10−3 10−4 10−5 10−6 10−7 10−10 Initial pH 1.08 1.62 2.25 3.05 4.01 5.00 5.97 6.74 7.00 Final pH 6.71 7.17 7.63 8.06 8.24 8.26 8.26 8.26 8.27 Dissolved CaCO3
(g/L of acid)62.0 7.39 0.874 0.123 0.0536 0.0477 0.0471 0.0471 0.0470
- where [A] = [H3PO4] + [H
2PO−
4] + [HPO2−
4] + [PO3−
4] is the total acid concentration. Thus phosphoric acid is more efficient than a monoacid since at the final almost neutral pH, the second dissociated state concentration [HPO2−
4] is not negligible (see phosphoric acid).
See also
- Cuttlebone
- Cuttlefish
- Gesso
- Limescale
- Marble
- Ocean acidification
- Whiting event
- List of climate engineering topics
- Lysocline
References
- "Occupational safety and health guideline for calcium carbonate" (PDF). US Dept. of Health and Human Services. Retrieved 31 March 2011.
- "Archived copy" (PDF). Archived from the original (PDF) on 29 October 2018. Retrieved 29 October 2018.CS1 maint: archived copy as title (link)
- Aylward, Gordon; Findlay, Tristan (2008). SI Chemical Data Book (4th ed.). John Wiley & Sons Australia. ISBN 978-0-470-81638-7.
- Rohleder, J.; Kroker, E. (2001). Calcium Carbonate: From the Cretaceous Period Into the 21st Century. Springer Science & Business Media. ISBN 978-3-7643-6425-0.
- Benjamin, Mark M. (2002). Water Chemistry. McGraw-Hill. ISBN 978-0-07-238390-4.
- Zumdahl, Steven S. (2009). Chemical Principles 6th Ed. Houghton Mifflin Company. p. A21. ISBN 978-0-618-94690-7.
- NIOSH Pocket Guide to Chemical Hazards. "#0090". National Institute for Occupational Safety and Health (NIOSH).
- "Precipitated Calcium Carbonate". Archived from the original on 11 January 2014. Retrieved 11 January 2014.
- Ropp, R. C. (6 March 2013). Encyclopedia of the Alkaline Earth Compounds. Elsevier. pp. 359–370. ISBN 9780444595508.
- Demichelis, Raffaella; Raiteri, Paolo; Gale, Julian D.; Dovesi, Roberto (2013). "The Multiple Structures of Vaterite". Crystal Growth & Design. 13 (6): 2247–2251. doi:10.1021/cg4002972. ISSN 1528-7483.
- Russell, Daniel E . 17 February 2008. Retrieved December 31, 2010. "Helgustadir Iceland Spar Mine" mindat.org
- Horne, Francis (23 October 2006). "How are seashells created?". Scientific American. Retrieved 25 April 2012.
- "Oyster shell calcium". WebMD. Retrieved 25 April 2012.
- "Oyster Shell Calcium Carbonate". Caltron Clays & Chemicals. Archived from the original on 10 September 2013. Retrieved 25 April 2012.
- Mangels, Ann Reed (4 June 2014). "Bone nutrients for vegetarians". The American Journal of Clinical Nutrition. 100 (1): 469S–475S. doi:10.3945/ajcn.113.071423. PMID 24898231.CS1 maint: date and year (link)
- Boynton, W. V.; Ming, D. W.; Kounaves, S. P.; et al. (2009). "Evidence for Calcium Carbonate at the Mars Phoenix Landing Site" (PDF). Science. 325 (5936): 61–64. Bibcode:2009Sci...325...61B. doi:10.1126/science.1172768. PMID 19574384.
- Clark, B. C., III; Arvidson, R. E.; Gellert, R.; et al. (2007). "Evidence for montmorillonite or its compositional equivalent in Columbia Hills, Mars" (PDF). Journal of Geophysical Research. 112 (E6): E06S01. Bibcode:2007JGRE..112.6S01C. doi:10.1029/2006JE002756. hdl:1893/17119.
- Weyl, P.K. (1959). "The change in solubility of calcium carbonate with temperature and carbon dioxide content". Geochimica et Cosmochimica Acta. 17 (3–4): 214–225. doi:10.1016/0016-7037(59)90096-1.
- Trexler, D. (2001). "Two Medicine Formation, Montana: geology and fauna". In Tanke, D. H.; Carpenter, K. (eds.). Mesozoic Vertebrate Life. Indiana University Press. pp. 298–309. ISBN 978-0-253-33907-2.
- Ward, Peter (2006). Out of Thin Air: Dinosaurs, Birds, and Earth's Ancient Atmosphere. doi:10.17226/11630. ISBN 9780309666121.
- "Effects of Acid Rain". US Environmental Protection Agency. Retrieved 14 March 2015.
- "Blast Furnace". Science Aid. Archived from the original on 17 December 2007. Retrieved 30 December 2007.
- Sfetcu, Nicolae (2 May 2014). Health & Drugs: Disease, Prescription & Medication. Nicolae Sfetcu.
- McGinnis, R. A. Beet-Sugar Technology (2nd ed.). Beet Sugar Development Foundation. p. 178.
- "Precipitated Calcium Carbonate uses". Archived from the original on 25 July 2014.
- "Market Study Fillers, 2nd ed". Ceresana. September 2011.
- "Calcium Carbonate Powder". Reade Advanced Materials. 4 February 2006. Archived from the original on 22 February 2008. Retrieved 30 December 2007.
- "Calcium carbonate in plastic applications". Imerys Performance Minerals. Archived from the original on 4 August 2008. Retrieved 1 August 2008.
- "Why do calcium carbonate play an important part in Industrial". www.xintuchemical.com. Retrieved 7 October 2018.
- "precipitated calcium carbonate commodity price". www.dgci.be. Archived from the original on 7 October 2018. Retrieved 7 October 2018.
- Jimoh, O.A.; et al. (2017). "Understanding the Precipitated Calcium Carbonate (PCC) Production Mechanism and Its Characteristics in the Liquid–Gas System Using Milk of Lime (MOL) Suspension" (PDF). South African Journal of Chemistry. 70: 1–7. doi:10.17159/0379-4350/2017/v70a1.
- "Ohio Historical Society Blog: Make It Shine". Ohio Historical Society. Archived from the original on 23 March 2012. Retrieved 2 June 2011.
- "Calcium Carbonate". Medline Plus. National Institutes of Health. 1 October 2005. Archived from the original on 17 October 2007. Retrieved 30 December 2007.
- Lieberman, Herbert A.; Lachman, Leon; Schwartz, Joseph B. (1990). Pharmaceutical Dosage Forms: Tablets. New York: Dekker. p. 153. ISBN 978-0-8247-8044-9.
- "Food Additives – Names Starting with C". Chemistry.about.com. 10 April 2012. Archived from the original on 16 October 2006. Retrieved 24 May 2012.
- Caruso JB, Patel RM, Julka K, Parish DC (July 2007). "Health-behavior induced disease: return of the milk-alkali syndrome". J Gen Intern Med. 22 (7): 1053–5. doi:10.1007/s11606-007-0226-0. PMC 2219730. PMID 17483976.
- Beall DP, Henslee HB, Webb HR, Scofield RH (May 2006). "Milk-alkali syndrome: a historical review and description of the modern version of the syndrome". Am. J. Med. Sci. 331 (5): 233–42. doi:10.1097/00000441-200605000-00001. PMID 16702792.
- Gabriely, Ilan; Leu, James P.; Barzel, Uriel S. (2008). "Clinical problem-solving, back to basics". New England Journal of Medicine. 358 (18): 1952–6. doi:10.1056/NEJMcps0706188. PMID 18450607.
- "E-numbers: E170 Calcium carbonate". Food-Info.net. 080419 food-info.net
- "Current EU approved additives and their E Numbers". UK Food Standards Agency. Retrieved 27 October 2011.
- "Listing of Food Additives Status Part I". US Food and Drug Administration. Archived from the original on 14 March 2013. Retrieved 27 October 2011.
- "Standard 1.2.4 – Labelling of ingredients". Australia New Zealand Food Standards Code. Retrieved 27 October 2011.
- Zhao, Y.; Martin, B. R.; Weaver, C. M. (2005). "Calcium bioavailability of calcium carbonate fortified soymilk is equivalent to cow's milk in young women". The Journal of Nutrition. 135 (10): 2379–2382. doi:10.1093/jn/135.10.2379. PMID 16177199.
- Oates, J. A. H. (11 July 2008). Lime and Limestone: Chemistry and Technology, Production and Uses. John Wiley & Sons. pp. 111–113. ISBN 978-3-527-61201-7.
- "Limestone Dispenser Fights Acid Rain in Stream". The New York Times. Associated Press. 13 June 1989.
- "Environmental Uses for Calcium Carbonate". Congcal. Retrieved 5 August 2013.
- Schreiber, R. K. (1988). "Cooperative federal-state liming research on surface waters impacted by acidic deposition". Water, Air, & Soil Pollution. 41 (1): 53–73. doi:10.1007/BF00160344 (inactive 16 March 2020).
- Kircheis, Dan; Dill, Richard (2006). "Effects of low pH and high aluminum on Atlantic salmon smolts in Eastern Maine and liming project feasibility analysis" (reprinted at Downeast Salmon Federation). National Marine Fisheries Service and Maine Atlantic Salmon Commission.
- Guhrén, M.; Bigler, C.; Renberg, I. (2006). "Liming placed in a long-term perspective: A paleolimnological study of 12 lakes in the Swedish liming program". Journal of Paleolimnology. 37 (2): 247–258. Bibcode:2007JPall..37..247G. doi:10.1007/s10933-006-9014-9.
- "Solvay Precipitated Calcium Carbonate: Production". Solvay. 9 March 2007. Retrieved 30 December 2007.
- Lide, D. R., ed. (2005). CRC Handbook of Chemistry and Physics (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- "Selected Solubility Products and Formation Constants at 25 °C". California State University, Dominguez Hills.
- Wang, X.; Conway, W.; Burns, R.; McCann, N.; Maeder, M. (2010). "Comprehensive Study of the Hydration and Dehydration Reactions of Carbon Dioxide in Aqueous Solution". The Journal of Physical Chemistry A. 114 (4): 1734–40. Bibcode:2010JPCA..114.1734W. doi:10.1021/jp909019u. PMID 20039712.
- Mook, W. (2000). "Chemistry of carbonic acid in water". Environmental Isotopes in the Hydrological Cycle: Principles and Applications (PDF). Paris: INEA/UNESCO. pp. 143–165.
- Wojtowicz, J. A. (1998). "Factors affecting precipitation of calcium carbonate" (PDF). Journal of the Swimming Pool and Spa Industry. 3 (1): 18–23. Archived from the original (PDF) on 18 March 2014. Retrieved 18 March 2014.
- Wojtowicz, J. A. (1998). "Corrections, potential errors, and significance of the saturation index" (PDF). Journal of the Swimming Pool and Spa Industry. 3 (1): 37–40. Archived from the original (PDF) on 24 August 2012. Retrieved 18 March 2014.
- Birch, R. G. (2013). "BABES: a better method than "BBB" for pools with a salt-water chlorine generator" (PDF). iinet.net.au.