Split gene theory
The "split gene" theory by Periannan Senapathy is a theory of the origin of introns, long non-coding sequences in eukaryotic genes that intervene the exons.[1][2][3] This theory holds that the randomness of primodial DNA sequences would only permit small (< 600bp) open reading frames, and that important intron structures and regulatory sequences are derived from stop codons. In this introns-first framework, the spliceosomal machinery and the nucleus evolved due to the necessity to join these ORFs (now "exons") into larger proteins, and that intronless bacterial genes are less ancestral than the split eukaryotic genes.
The theory provides solutions to the key questions concerning the split eukaryotic genes, including the exons, introns, splice junctions, branch points and the entire split gene architecture, based on the origin of split genes from random genetic sequences. It also provides possible solutions to the origin of the spliceosomal machinery, the nuclear boundary and the eukaryotic cell. This theory led to the Shapiro–Senapathy algorithm, which provides the methodology for detecting the splice sites, exons and split genes in a eukaryotic DNA, and which is the main method for detecting splice site mutations in genes that cause hundreds of diseases in thousands of patients worldwide.
The details of how the split gene theory was formulated, and how this theory is corroborated in every aspect of the genetic elements of the eukaryotic gene by the published literature, are provided below.
Split gene theory requires a separate origin of all eukaryotic species. It also requires that the simpler prokaryotes have evolved from eukaryotes. This completely contradicts the scientific consensus about the formation of eukaryotic cells by endosymbiosis of bacteria. In 1994, Senapathy wrote a book about this aspect of his theory - The Independent Birth of Organisms. It proposed that all eukaryotic genomes were formed separately in a primordial pool. The Dutch biologist Gert Korthoff criticized the theory by posing various problems which can't be explained by a theory of independent origins. He also pointed out that various eukaryotes need parental care and called this the 'boot problem'. There couldn't have been any first ancestor who needed parental care. Korthoff notes that a large fraction of eukaryotes are parasites. Senapathy's theory would require a coincidence to explain their existence. Senapathy's theory also cannot explain the strong evidence for common descent (like homology, universal genetic code, embryology, fossil record.)[4]
Background
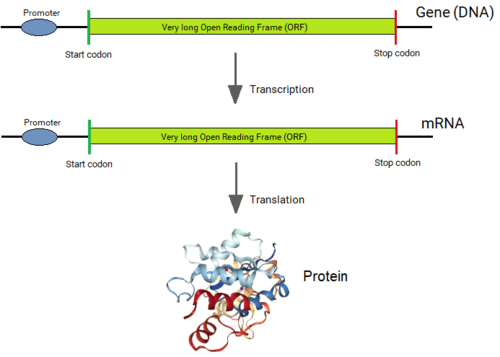
Genes of all organisms, except bacteria, consist of short protein-coding regions (exons) interrupted by long sequences that intervene the coding sequences (introns).[1][2] When a gene is expressed, its DNA sequence is copied into a “primary RNA” sequence by the enzyme RNA polymerase. Then the “spliceosome” machinery physically removes the introns from the RNA copy of the gene by the process of splicing, leaving only a contiguously connected series of exons, which becomes the “messenger” RNA (mRNA). This mRNA is now “read” by another cellular machinery, called the “ribosome,” to produce the encoded protein. Thus, although introns are not physically removed from a gene, a gene's sequence is read as if introns never existed.
The exons are usually very short, with an approx. average length of about 120 bases (e.g. in human genes). The length of introns varies widely between 10 bases to 500,000 bases in a genome (for example, the human genome), but the length of exons has an upper limit of about 600 bases in most of the eukaryotic genes. Because exons code for protein sequences, they are very important for the cell, yet constitute only ~2% of the genes’ sequences. Introns, in contrast, constitute 98% of the genes’ sequences but seem to have little crucial functions in genes, except for functions such as containing enhancer sequences and developmental regulators in rare instances.[5][6]
Until Philip Sharp [7][8] from the MIT and Richard Roberts [9] then at the Cold Spring Harbor Laboratories (currently at the New England Biolabs) discovered introns[10] within eukaryotic genes in 1977, it was believed that the coding sequence of all genes was always in one single stretch, bounded by a single long Open Reading Frame (ORF). The discovery of introns was a profound surprise to scientists, which instantly brought up the questions of how, why and when the introns came into the eukaryotic genes.
It soon became apparent that a typical eukaryotic gene was interrupted at many locations by introns, dividing the coding sequence into many short exons. Also surprising was that the introns were very long, even as long as hundreds of thousands of bases (see table below). These findings also prompted the questions of why many introns occur within a gene (for example, ~312 introns occur in the human gene TTN), why they are very long, and why exons are very short.
| Gene symbol | Gene length (bases) |
Longest Intron length (bases) |
Number of
introns in the gene |
|---|---|---|---|
| ROBO2 | 1,743,269 | 1,160,411 | 104 |
| KCNIP4 | 1,220,183 | 1,097,903 | 76 |
| ASIC2 | 1,161,877 | 1,043,911 | 18 |
| NRG1 | 1,128,573 | 956,398 | 177 |
| DPP10 | 1,403,453 | 866,399 | 142 |
| The longest introns in the human genes. | |||
It was also discovered that the spliceosome machinery was very large and complex with ~300 proteins and several SnRNA molecules. So, the questions also extended to the origin of the spliceosome. Soon after the discovery of introns, it became apparent that the junctions between exons and introns on either side exhibited specific sequences that signalled the spliceosome machinery to the exact base position for splicing. How and why these splice junction signals came into being was another important question to be answered.
Early speculations
The startling discovery of introns and the split gene architecture of the eukaryotic genes was dramatic, and started a new era of eukaryotic biology. The question of why eukaryotic genes had a genes-in-pieces architecture prompted speculations and discussions in the literature almost immediately.
Ford Doolittle from the Dalhousie University published a paper in 1978 in which he expressed his views.[11] He stated that most molecular biologists assumed that the eukaryotic genome arose from a ‘simpler’ and more ‘primitive’ prokaryotic genome rather like that of Escherichia coli. However, this type of evolution would require that introns be introduced into the contiguous coding sequences of bacterial genes. Regarding this requirement, Doolittle said, “It is extraordinarily difficult to imagine how informationally irrelevant sequences could be introduced into pre-existing structural genes without deleterious effects.” He stated “I would like to argue that the eukaryotic genome, at least in that aspect of its structure manifested as ‘genes in pieces’ is in fact the primitive original form.”
James Darnell from the Rockefeller University also expressed similar views in 1978.[12] He stated, “The differences in the biochemistry of messenger RNA formation in eukaryotes compared to prokaryotes are so profound as to suggest that sequential prokaryotic to eukaryotic cell evolution seems unlikely. The recently discovered non-contiguous sequences in eukaryotic DNA that encode messenger RNA may reflect an ancient, rather than a new, distribution of information in DNA and that eukaryotes evolved independently of prokaryotes.”
However, in an apparent attempt to reconcile with the idea that RNA preceded DNA in evolution, and with the concept of the three evolutionary lineages of archea, bacteria and eukarya, both Doolittle and Darnell deviated from their original speculation in a paper they published together in 1985.[13] They suggested that the ancestor of all three groups of organisms, the ‘progenote,’ had a genes-in-pieces structure, from which all three lineages evolved. They speculated that the precellular stage had primitive RNA genes which had introns, which were reverse transcribed into DNA and formed the progenote. Bacteria and archea evolved from the progenote by losing introns, and ‘urkaryote’ evolved from it by retaining introns. Later, the eukaryote evolved from the urkaryote by evolving a nucleus and gaining the mitochondria from the bacteria. Multicellular organisms then evolved from the eukaryote.
These authors were able to predict that the distinctions between the prokaryote and the eukaryote were so profound that the prokaryote to eukaryote evolution was not tenable, and that both had different origins. However, other than the speculations that the precellular RNA genes must have had introns, they did not address the key questions of where from, how or why the introns could have originated in these genes or what their material basis was. There were no explanations of why exons were short and introns were long, how the splice junctions originated, what the structure and sequence of the splice junctions meant, and why eukaryotic genomes were large.
Around the same time that Doolittle and Darnell suggested that introns in eukaryotic genes could be ancient, Colin Blake[14] from the university of Oxford and Walter Gilbert[15][16] from Harvard University (who won the Nobel Prize for inventing a DNA sequencing method along with Fred Sanger) published their views on intron origins independently. In their view, introns originated as spacer sequences that enabled the recombination and shuffling of exons that encoded distinct functional domains in order to evolve new genes. Thus, new genes were assembled from exon modules that coded for functional domains, folding regions, or structural elements from preexisting genes in the genome of an ancestral organism, thereby evolving genes with new functions. They did not specify how the exons representing protein structural motifs originated, or the introns that do not code for proteins originated. In addition, even after many years, extensive analysis of several thousands of proteins and genes showed that only extremely rarely do genes exhibit the supposed exon shuffling phenomenon.[17][18] Furthermore, several molecular biologists had questioned the exon shuffling proposal, from a purely evolutionary view for both methodological and conceptual reasons, and, in the long run, this theory did not materialize.
Hypothesis
Around the same time introns were discovered, Senapathy was asking how genes themselves could have originated. He surmised that for any gene to come into being, there must have been genetic sequences (RNA or DNA) present in the prebiotic chemistry environment. A basic question he asked was how protein-coding sequences could have originated from primordial DNA sequences at the initial development of the very first cells.
To answer this, he made two basic assumptions: (i) before a self-replicating cell could come into existence, DNA molecules were synthesized in the primordial soup by random addition of the 4 nucleotides without the help of templates and (ii) the nucleotide sequences that code for proteins were selected from these preexisting random DNA sequences in the primordial soup, and not by construction from shorter coding sequences. He also surmised that codons must have been established prior to the origin of the first genes. If primordial DNA did contain random nucleotide sequences, he asked: Was there an upper limit in the coding-sequence lengths, and, if so, did this limit play a crucial role in the formation of the structural features of genes at the very beginning of the origin of genes?
His logic was the following. The average length of proteins in living organisms, including the eukaryotic and bacterial organisms, was ~400 amino acids. However, there existed much longer proteins, even longer than 10,000 amino acids up to ~30,000 amino acids, in both eukaryotes and bacteria.[19] Thus, the coding sequence of thousands of bases existed in a single stretch in bacterial genes. In contrast, the coding sequence of eukaryotes existed only in short segments of exons of approx. 120 bases regardless of the length of the protein. If the coding sequence ORF lengths in random DNA sequences were as long as those in bacterial organisms, then contiguously long coding genes were possible to have occurred in random DNA. This was not known, as the distribution of the lengths of ORFs in a random DNA sequence was never studied before.
As random DNA sequences could be generated in the computer, Senapathy thought that he could ask these questions and conduct his experiments in the computer. Furthermore, when he began studying this question, there existed just about sufficient amount of DNA and protein sequence information in the National Biomedical Research Foundation (NBRF) database in the early 1980s.
Testing the hypothesis
Origin of introns and the split gene structure
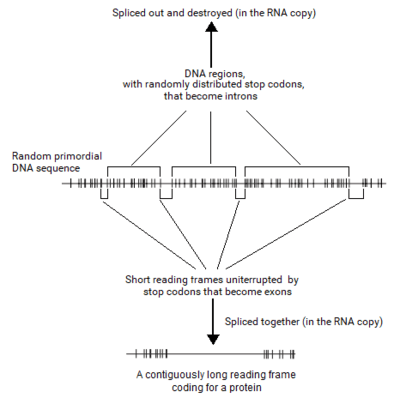
Senapathy analyzed the distribution of the ORF lengths in computer-generated random DNA sequences first. Surprisingly, this study revealed that there actually existed an upper limit of about 200 codons (600 bases) in the lengths of ORFs. The shortest ORF (zero base in length) was the most frequent. At increasing lengths of ORFs, their frequency decreased logarithmically, reaching almost zero at about 600 bases. When the probability of ORF lengths in a random sequence was plotted, it also revealed that the probability of increasing lengths of ORFs decreased exponentially and tailed off at a maximum of about 600 bases. From this “negative exponential” distribution of ORF lengths, it was found that most of the ORFs were extremely shorter than even the maximum of 600 bases.
This finding was surprising because the coding sequence for the average protein length of 400 AAs (with ~1,200 bases of coding sequence) and longer proteins of thousands of AAs (requiring >10,000 bases of coding sequence) would not occur at a stretch in a random sequence. If this was true, a typical gene with a contiguous coding sequence could not originate in a random sequence. Thus, the only possible way that any gene could originate from a random sequence was to split the coding sequence into shorter segments and select these segments from short ORFs available in the random sequence, rather than to increase the length of an ORF by eliminating numerous consecutively occurring stop codons. This process of choosing short segments of coding sequences from the available ORFs to make a long ORF would lead to a split structure of the gene.
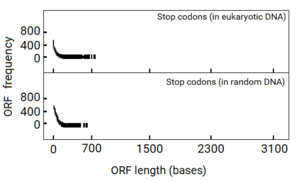
If this hypothesis was true, eukaryotic DNA sequences should show evidence for it. When Senapathy plotted the distribution of ORF lengths in eukaryotic DNA sequences, the plot was remarkably similar to that from random DNA sequence. This plot was also a negative exponential distribution that tailed off at a maximum of about 600 bases. This finding was amazing because the exons from eukaryotic genes also exhibited a maximum length of about 600 bases,[1][20][3] which coincided exactly with the maximum length of ORFs observed in both random DNA sequence and in eukaryotic DNA sequence.
The split genes thus originated from random DNA sequences by choosing the best of the short coding segments (exons) and joining them by a process of splicing. The intervening intron sequences were left-over vestiges of the random sequences, and thus were earmarked to be removed by the spliceosome. These findings indicated that split genes could have originated from random DNA sequences with exons and introns as they are found in today's eukaryotic organisms. The Nobel Laureate Marshall Nirenberg, who deciphered the codons, stated that these findings strongly showed that the split gene theory for the origin of introns and the split structure of genes must be valid.[1] New Scientist covered this publication in “A long explanation for introns”.[21]
Noted molecular biologist Dr. Colin Blake from the university of Oxford, who proposed the Gilbert-Blake hypothesis in 1979 for the origin of introns (see above), stated in his 1987 publication entitled “Proteins, exons and molecular evolution,” that Senapathy's split gene theory comprehensively explained the origin of the split gene structure. In addition, he stated that it explained several key questions including the origin of the splicing mechanism:[14]
“Recent work by Senapathy, when applied to RNA, comprehensively explains the origin of the segregated form of RNA into coding and non-coding regions. It also suggests why a splicing mechanism was developed at the start of primordial evolution. He found that the distribution of reading frame lengths in a random nucleotide sequence corresponded exactly to that for the observed distribution of eukaryotic exon sizes. These were delimited by regions containing stop signals, the messages to terminate construction of the polypeptide chain, and were thus non-coding regions or introns. The presence of a random sequence was therefore sufficient to create in the primordial ancestor the segregated form of RNA observed in the eukaryotic gene structure. Moreover, the random distribution also displays a cutoff at 600 nucleotides, which suggests that the maximum size for an early polypeptide was 200 residues, again as observed in the maximum size of the eukaryotic exon. Thus, in response to evolutionary pressures to create larger and more complex genes, the RNA fragments were joined together by a splicing mechanism that removed the introns. Hence, the early existence of both introns and RNA splicing in eukaryotes appears to be very likely from a simple statistical basis. These results also agree with the linear relationship found between the number of exons in the gene for a particular protein and the length of the polypeptide chain.”

Origin of splice junctions
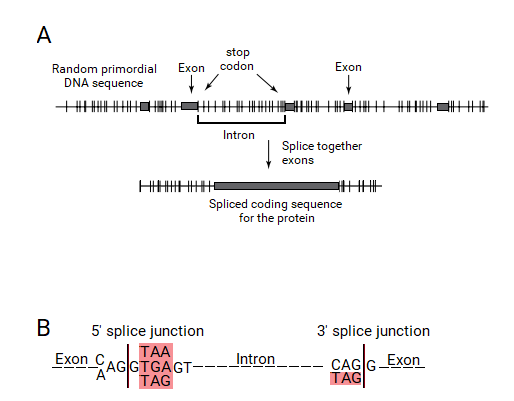
Under the split gene theory, an exon would be defined by an ORF. It would require that a mechanism to recognize an ORF should have originated. As an ORF is defined by a contiguously coding sequence bounded by stop codons, these stop codon ends had to be recognized by this exon-intron gene recognition system. This system could have defined the exons by the presence of a stop codon at the ends of ORFs, which should be included within the ends of the introns and eliminated by the splicing process. Thus, the introns should contain a stop codon at their ends, which would be part of the splice junction sequences.
If this hypothesis was true, the split genes of today's living organisms should contain stop codons exactly at the ends of introns. When Senapathy tested this hypothesis in the splice junctions of eukaryotic genes, it was astonishing that the vast majority of splice junctions did contain a stop codon at the ends of introns, right outside of the exons. In fact, these stop codons were found to form the “canonical” GT:AG splicing sequence, with the three stop codons occurring as part of the strong consensus signals. Thus, the basic split gene theory for the origin of introns and the split gene structure led to the understanding that the splice junctions originated from the stop codons.[2]
| Codon | Number of occurrences in donor signal |
Number of occurrences in acceptor signal |
|---|---|---|
| TAA | 370 | 0 |
| TGA | 293 | 0 |
| TAG | 64 | 234 |
| CAG | 7 | 746 |
| Other | 297* | 50 |
| Total | 1030 | 1030 |
| Frequency of stop codons in donor and acceptor splice-junction sequences.[2]
Sequence data for only about 1,000 exon-intron junctions were available when *More than 70% are TAX [TAT = 75; TAC = 59; TGT = 70]. | ||
All three stop codons (TGA, TAA and TAG) were found after one base (G) at the start of introns. These stop codons are shown in the consensus canonical donor splice junction as AG:GT(A/G)GGT, wherein the TAA and TGA are the stop codons, and the additional TAG is also present at this position. Besides the codon CAG, only TAG, which is a stop codon, was found at the ends of introns. The canonical acceptor splice junction is shown as (C/T)AG:GT, in which TAG is the stop codon. These consensus sequences clearly show the presence of the stop codons at the ends of introns bordering the exons in all eukaryotic genes, thus providing a strong corroboration for the split gene theory. Marshall Nirenberg again stated that these observations fully supported the split gene theory for the origin of splice junction sequences from stop codons, who was the referee for this paper.[2] New Scientist covered this publication in “Exons, Introns and Evolution”.[22]
Soon after the discovery of introns by Drs. Philip Sharp and Richard Roberts, it became known that mutations within splice junctions could lead to diseases. Senapathy showed that mutations in the stop codon bases (canonical bases) caused more diseases than the mutations in non-canonical bases.[1]
Branch point (lariat) sequence
An intermediate stage in the process of eukaryotic RNA splicing is the formation of a lariat structure. It is anchored at an adenosine residue in intron between 10 and 50 nucleotides upstream of the 3' splice site. A short conserved sequence (the branch point sequence) functions as the recognition signal for the site of lariat formation. During the splicing process, this conserved sequence towards the end of the intron forms a lariat structure with the beginning of the intron.[23] The final step of the splicing process occurs when the two exons are joined and the intron is released as a lariat RNA.[24]
Several investigators have found the branch point sequences in different organisms[23] including yeast, human, fruit fly, rat, and plants. Senapathy found that, in all of these branch point sequences, the codon ending at the branch point adenosine is consistently a stop codon. What is interesting is that two of the three stop codons (TAA and TGA) occur almost all of the times at this position.
| Organism | Lariat Consensus sequence |
|---|---|
| Yeast | TACTAAC |
| Human Beta globin genes | CTGAC
CTAAT CTGAT CTAAC CTCAC |
| Drosophila | CTAAT |
| Rats | CTGAC |
| Plants | (C/T)T(A/G)A(T/C) |
| Consistent presence of stop codons in branch point signal sequences.
Lariat (branch point) sequences have been identified from many different organisms.These sequences consistently show that the codon ending in the branching adenosine is a stop codon, either TAA or TGA, which are shown in red. | |
These findings led Senapathy to propose that the branch point signal originated from stop codons. The finding that two different stop codons (TAA and TGA) occur within the lariat signal with the branching point as the third base of the stop codons corroborates this proposal. As the branching point of the lariat occurs at the last adenine of the stop codon, it is possible that the spliceosome machinery that originated for the elimination of the numerously occurring stop codons from the primary RNA sequence created an auxiliary stop-codon sequence signal as the lariat sequence to aid its splicing function.[2]
The small nuclear U2 RNA found in splicing complexes is thought to aid splicing by interacting with the lariat sequence.[25] Complementary sequences for both the lariat sequence and the acceptor signal are present in a segment of only 15 nucleotides in U2 RNA. Further, the U1 RNA has been proposed to function as a guide in splicing to identify the precise donor splice junction by complementary base-pairing. The conserved regions of the U1 RNA thus include sequences complementary to the stop codons. These observations enabled Senapathy to predict that stop codons had operated in the origin of not only the splice-junction signals and the lariat signal, but also some of the small nuclear RNAs.
Gene regulatory sequences
Dr Senapathy also proposed that the gene-expression regulatory sequences (promoter and poly-A addition site sequences) also could have originated from stop codons. A conserved sequence, AATAAA, exists in almost every gene a short distance downstream from the end of the protein-coding message and serves as a signal for the addition of poly(A) in the mRNA copy of the gene.[26] This poly(A) sequence signal contains a stop codon, TAA. A sequence shortly downstream from this signal, thought to be part of the complete poly(A) signal, also contains the TAG and TGA stop codons.
Eukaryotic RNA-polymerase-II-dependent promoters can contain a TATA box (consensus sequence TATAAA), which contains the stop codon TAA. Bacterial promoter elements at -10 bases exhibits a TATA box with a consensus of TATAAT (which contains the stop codon TAA), and at -35 bases exhibits a consensus of TTGACA (containing the stop codon TGA). Thus, the evolution of the whole RNA processing mechanism seems to have been influenced by the too-frequent occurrence of stop codons in DNA sequence, thus making the stop codons the focal points for RNA processing.
Stop codons are key parts of every genetic element in the eukaryotic gene
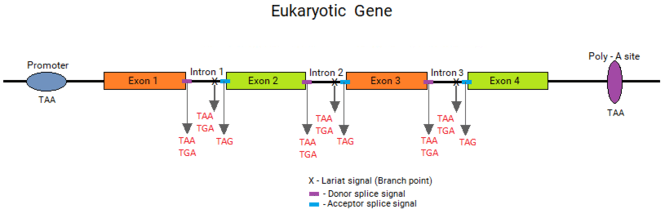
| Genetic Element | Consensus sequence |
|---|---|
| Promoter | TATAAT |
| Donor Splice Sequence | CAG:GTAAGT
CAG:GTGAGT |
| Acceptor Splice Sequence | (C/T)9...TAG:GT |
| Lariat Sequence | CTGAC
CTAAC |
| Poly-A addition site | TATAAA |
| The consistent occurrence of stop codons in genetic elements in eukaryotic genes.The consensus sequences of the different genetic elements in eukaryotic genes are shown. The stop codon(s) in each of these sequences are colored in red. | |
Dr. Senapathy's work based on his split gene theory has unraveled that stop codons occur as the key parts in every genetic element in eukaryotic genes. The table and figure above show that the key parts of the core promoter elements, the lariat (branch point) signal, the donor and acceptor splice signals, and the poly-A addition signal consist of one or more stop codons. This finding provides a strong corroboration for the split gene theory that the underlying reason for the complete split gene paradigm is the origin of split genes from random DNA sequences, wherein random distribution of an extremely high frequency of stop codons were used by nature to define these genetic elements.
Why exons are short and introns are long?
Research based on the split gene theory sheds light on other basic questions of exons and introns. The exons of eukaryotes are generally short (human exons average ~120 bases, and can be as short as 10 bases) and introns are usually very long (average of ~3,000 bases, and can be several hundred thousands bases long), for example genes RBFOX1, CNTNAP2, PTPRD and DLG2. Senapathy has provided a plausible answer to these questions, which has remained the only explanation so far. Based on the split gene theory, exons of eukaryotic genes, if they originated from random DNA sequences, have to match the lengths of ORFs from random sequence, and possibly should be around 100 bases (close to the median length of ORFs in random sequence). The genome sequences of living organisms, for example the human, exhibits exactly the same average lengths of 120 bases for exons, and the longest exons of 600 bases (with few exceptions), which is the same length as that of the longest random ORFs.[1][2][3][20]
If split genes originated in random DNA sequences, then introns would be long for several reasons. The stop codons occur in clusters leading to numerous consecutive very short ORFs, and longer ORFs that could be defined as exons would be rarer. Furthermore, the best of the coding sequence parameters for functional proteins would be chosen from the long ORFs in random sequence, which may occur rarely. In addition, the combination of the donor and acceptor splice junction sequences within short lengths of coding sequence segments that would define exon boundaries would occur rarely in a random sequence. These combined reasons would make introns very long compared to the lengths of exons.
Why eukaryotic genomes are large?
This work also explains why the genomes are very large, for example, the human genome with three billion bases, and why only a very small fraction of the human genome (~2%) codes for the proteins and other regulatory elements.[27][28] If split genes originated from random primordial DNA sequences, it would contain a significant amount of DNA that would be represented by introns. Furthermore, a genome assembled from random DNA containing split genes would also include intergenic random DNA. Thus, the nascent genomes that originated from random DNA sequences had to be large, regardless of the complexity of the organism.
The observation that the genomes of several organisms such as that of the onion (~16 billion bases[29]) and salamander (~32 billion bases[30]) are much larger than that of the human (~3 billion bases[31][32]) but the organisms are no more complex than human provides credence to this split gene theory. Furthermore, the findings that the genomes of several organisms are smaller, although they contain essentially the same number of genes as that of the human, such as those of the C. elegans (genome size ~100 million bases, ~19,000 genes)[33] and Arabidopsis thaliana (genome size ~125 million bases, ~25,000 genes),[34] adds support to this theory. The split gene theory predicts that the introns in the split genes in these genomes could be the “reduced” (or deleted) form compared to the larger genes with long introns, thus leading to reduced genomes.[1][20] In fact, researchers have recently proposed that these smaller genomes are actually reduced genomes, which adds support to the split gene theory.[35]
Origin of the spliceosomal machinery and eukaryotic nucleus
Senapathy's research also addresses the origin of the spliceosomal machinery that edits out the introns from the RNA transcripts of genes. If the split genes had originated from random DNA, then the introns would have become an unnecessary but integral part of the eukaryotic genes along with the splice junctions at their ends. The spliceosomal machinery would be required to remove them and to enable the short exons to be linearly spliced together as a contiguously coding mRNA that can be translated into a complete protein. Thus, the split gene theory shows that the whole spliceosomal machinery originated due to the origin of split genes from random DNA sequences, and to remove the unnecessary introns.[1][2]
As noted above, Colin Blake, the author of the Gilbert-Blake theory for the origin of introns and exons, states, “Recent work by Senapathy, when applied to RNA, comprehensively explains the origin of the segregated form of RNA into coding and noncoding regions. It also suggests why a splicing mechanism was developed at the start of primordial evolution.”[14]
Senapathy had also proposed a plausible mechanistic and functional rationale why the eukaryotic nucleus originated, a major question in biology.[1][2] If the transcripts of the split genes and the spliced mRNAs were present in a cell without a nucleus, the ribosomes would try to bind to both the un-spliced primary RNA transcript and the spliced mRNA, which would result in a molecular chaos. If a boundary had originated to separate the RNA splicing process from the mRNA translation, it can avoid this problem of molecular chaos. This is exactly what is found in eukaryotic cells, where the splicing of the primary RNA transcript occurs within the nucleus, and the spliced mRNA is transported to the cytoplasm, where the ribosomes translate them into proteins. The nuclear boundary provides a clear separation of the primary RNA splicing and the mRNA translation.
Origin of the eukaryotic cell
These investigations thus led to the possibility that primordial DNA with essentially random sequence gave rise to the complex structure of the split genes with exons, introns and splice junctions. They also predict that the cells that harbored these split genes had to be complex with a nuclear cytoplasmic boundary, and must have had a spliceosomal machinery. Thus, it was possible that the earliest cell was complex and eukaryotic.[1][2][3][20] Surprisingly, findings from extensive comparative genomics research from several organisms over the past 15 years are showing overwhelmingly that the earliest organisms could have been highly complex and eukaryotic, and could have contained complex proteins,[36][37][38][39][40][41][42] exactly as predicted by Senapathy's theory.
The spliceosome is a highly complex machinery within the eukaryotic cell, containing ~200 proteins and several SnRNPs. In their paper [43] “Complex spliceosomal organization ancestral to extant eukaryotes,” molecular biologists Lesley Collins and David Penny state “We begin with the hypothesis that ... the spliceosome has increased in complexity throughout eukaryotic evolution. However, examination of the distribution of spliceosomal components indicates that not only was a spliceosome present in the eukaryotic ancestor but it also contained most of the key components found in today's eukaryotes. ... the last common ancestor of extant eukaryotes appears to show much of the molecular complexity seen today.” This suggests that the earliest eukaryotic organisms were highly complex and contained sophisticated genes and proteins, as the split gene theory predicts.
Origin of bacterial genes
Based on the split gene theory, only genes split into short exons and long introns, with a maximum exon length of ~600 bases, could have occurred in random DNA sequences. Genes with long uninterrupted coding sequences that are thousands of bases long and longer than 10,000 bases up to 90,000 bases that occur in many bacterial organisms[19] were practically impossible to have occurred. However, the bacterial genes could have originated from split genes by losing introns, which seems to be the only way to arrive at long coding sequences. It is also a better way than by increasing the lengths of ORFs from very short random ORFs to very long ORFs by specifically removing the stop codons by mutation.[1][2][3]
| Gene size (bases) | Number of genes |
|---|---|
| 5,000 - 10,000 | 3,029 |
| 10,000 - 15,000 | 492 |
| 15,000 - 20,000 | 131 |
| 20,000 - 25,000 | 39 |
| >25,000 | 41 |
| Extremely long coding sequences occur as very long ORFs in bacterial genes. Thousands of genes that are longer than 5,000 bases, coding for proteins that are longer than 2,000 amino acids, exist in many bacterial genomes. The longest genes are ~90,000 bases long coding for proteins ~30,000 amino acids long. Each of these genes occur in a single stretch of coding sequence (ORF) without any interrupting stop codons or intervening introns. Data taken from Think big – giant genes in bacteria.[19] | |
According to the split gene theory, this process of intron loss could have happened from prebiotic random DNA. These contiguously coding genes could be tightly organized in the bacterial genomes without any introns and be more streamlined. According to Senapathy, the nuclear boundary that was required for a cell containing split genes in its genome (see the section Origin of the eukaryotic cell nucleus, above) would not be required for a cell containing only contiguously coding genes. Thus, the bacterial cells did not develop a nucleus. Based on split gene theory, the eukaryotic genomes and bacterial genomes could have independently originated from the split genes in primordial random DNA sequences.
The Shapiro-Senapathy algorithm
Based on the split gene theory, Senapathy developed computational algorithms to detect the donor and acceptor splice sites, exons and a complete split gene in a genomic sequence. He developed the position weight matrix (PWM) method based on the frequency of the four bases at the consensus sequences of the donor and acceptor in different organisms to identify the splice sites in a given sequence. Furthermore, he formulated the first algorithm to find the exons based on the requirement of exons to contain a donor sequence (at the 5’ end) and an acceptor sequence (at the 3’ end), and an ORF in which the exon should occur, and another algorithm to find a complete split gene. These algorithms are collectively known as the Shapiro-Senapathy algorithm (S&S).[44][45]
This Shapiro-Senapathy algorithm aids in the identification of splicing mutations that cause numerous diseases and adverse drug reactions.[44][45] Using the S&S algorithm, scientists have identified mutations and genes that cause numerous cancers, inherited disorders, immune deficiency diseases and neurological disorders (see here for details). It is increasingly used in clinical practice and research not only to find mutations in known disease-causing genes in patients, but also to discover novel genes that are causal of different diseases. Furthermore, it is used in defining the cryptic splice sites and deducing the mechanisms by which mutations in them can affect normal splicing and lead to different diseases. It is also employed in addressing various questions in basic research in humans, animals and plants.
The widespread use of this algorithm in biological research and clinical applications worldwide adds credence to the split gene theory, as this algorithm emanated from the split gene theory. The findings based on S&S have impacted major questions in eukaryotic biology and their applications to human medicine. These applications may expand as the fields of clinical genomics and pharmacogenomics magnify their research with mega sequencing projects such as the All of Us project[46] that will sequence a million individuals, and with the sequencing of millions of patients in clinical practice and research in the future.
Corroborating evidence
If the split gene theory is correct, the structural features of split genes predicted from computer-simulated random sequences can be expected to occur in actual eukaryotic split genes. This is what we find in most known split genes in eukaryotes living today. The eukaryotic sequences exhibit a nearly perfect negative exponential distribution of ORFs lengths, with an upper limit of 600 bases (with rare exceptions).[1][2][20][3] Also, with rare exceptions, the exons of eukaryotic genes fall within this 600 bases upper maximum.
Moreover, if this theory is correct, exons should be delimited by stop codons, especially at the 3’ ends of exons (that is, the 5’ end of introns). Actually they are precisely delimited more strongly at the 3’ ends of exons and less strongly at the 5’ ends in most known genes, as predicted.[1][2][20][3] These stop codons are the most important functional parts of both splice junctions (the canonical bases GT:AG). The theory thus provides an explanation for the “conserved” splice junctions at the ends of exons and for the loss of these stop codons along with introns when they are spliced out. If this theory is correct, splice junctions should be randomly distributed in eukaryotic DNA sequences, and they are.[3][23][44][45] The splice junctions present in transfer RNA genes and ribosomal RNA genes, which do not code for proteins and wherein stop codons have no functional meaning, should not contain stop codons, and again, this is observed. The lariat signal, another sequence involved in the splicing process, also contains stop codons.[1][2][3][20][23][44][45]
If the Split Gene theory is true, then introns should be non-coding. This is exactly found to be true in eukaryotic organisms living today, even when introns are hundreds of thousands of bases long. They should also be mostly non-functional, and they are. Except for some intron sequences including the donor and acceptor splice signal sequences and branch point sequences, and possibly the intron splice enhancers that occur at the ends of introns, which aid in the removal of introns, the vast majority of introns are devoid of any functions. The Split Gene theory does not preclude for rare sequences within introns to fortuitously exhibit functional elements that could be used by the genome and the cell, especially because the introns are extremely long, which is found to be true. All of these findings show that the predictions of the split gene theory are precisely corroborated by the structural and functional characteristics of the major genetic elements in split genes in modern eukaryotic organisms.
If the split genes originated from random primordial DNA sequences, as proposed in the split gene theory, there could be evidence that they were present in the earliest organisms. Actually, using comparative analysis of the modern genome data from several living organisms, scientists have found that the characteristics of split genes that are present in modern eukaryotes trace back to the earliest organisms that came on earth. These studies show that the earliest organisms could have contained the intron-rich split genes and complex proteins that occur in today's living organisms.[47][48][49][50][51][52][53][54][55]
In addition, using another computational analytical method known as the “maximum likelihood analysis,” scientists have found that the earliest eukaryotic organisms must have contained the same genes from today's living organisms with even a higher density of introns.[56] Furthermore, comparative genomics of many organisms including basal eukaryotes (considered to be primitive eukaryotic organisms such as Amoeboflagellata, Diplomonadida, and Parabasalia) have shown that intron-rich split genes accompanied by a fully formed spliceosome from today's complex organisms were present in the earliest organisms, and that the earliest organisms were extremely complex with all of the eukaryotic cellular components.[57][47][58][59][60][56]
These findings from the literature are exactly as predicted by the split gene theory, almost to a mathematical precision, providing remarkable support. This theory is corroborated by the findings from comparative analysis of actual eukaryotic gene sequences with those of the computer generated random DNA sequences. Furthermore, comparative analysis of genome data from many organisms living today by several groups of scientists show that the earliest organisms that appeared on earth had intron-rich split genes, coding for complex proteins and cellular components, such as those found in the modern eukaryotic organisms. Thus, the split gene theory provides comprehensive solutions to the entire structural and functional features of the split gene architecture, with strong corroborating evidence from published literature.
Selected publications
- Shapiro, Marvin B.; Senapathy, Periannan (1987). "RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression". Nucleic Acids Research. 15 (17): 7155–7174. doi:10.1093/nar/15.17.7155. PMC 306199. PMID 3658675.
- Senapathy, P. (1988). "Possible evolution of splice-junction signals in eukaryotic genes from stop codons". Proc Natl Acad Sci U S A. 85 (4): 1129–33. Bibcode:1988PNAS...85.1129S. doi:10.1073/pnas.85.4.1129. PMC 279719. PMID 3422483.
- Senapathy, P; Shapiro, MB; Harris, NL (1990). Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods in Enzymology. 183. pp. 252–78. doi:10.1016/0076-6879(90)83018-5. ISBN 9780121820848. PMID 2314278.
- Harris, N.L.; Senapathy, P. (1990). "Distribution and consensus of branch point signals in eukaryotic genes: a computerized statistical analysis". Nucleic Acids Res. 18 (10): 3015–9. doi:10.1093/nar/18.10.3015. PMC 330832. PMID 2349097.
- Senapathy, P. (1986). "Origin of eukaryotic introns: a hypothesis, based on codon distribution statistics in genes, and its implications". Proc Natl Acad Sci U S A. 83 (7): 2133–7. Bibcode:1986PNAS...83.2133S. doi:10.1073/pnas.83.7.2133. PMC 323245. PMID 3457379.
- Regulapati, R.; Bhasi, A.; Singh, C.K.; Senapathy, P. (2008). "Origination of the Split Structure of Spliceosomal Genes from Random Genetic Sequences". PLoS ONE. 3 (10): 10. Bibcode:2008PLoSO...3.3456R. doi:10.1371/journal.pone.0003456. PMC 2565106. PMID 18941625.
- Senapathy, P. (1995). "Introns and the origin of protein-coding genes". Science. 268 (5215): 1366–7. Bibcode:1995Sci...268.1366S. doi:10.1126/science.7761858. PMID 7761858.
References
- Senapathy, P. (April 1986). "Origin of eukaryotic introns: a hypothesis, based on codon distribution statistics in genes, and its implications". Proceedings of the National Academy of Sciences of the United States of America. 83 (7): 2133–2137. Bibcode:1986PNAS...83.2133S. doi:10.1073/pnas.83.7.2133. ISSN 0027-8424. PMC 323245. PMID 3457379.
- Senapathy, P. (February 1982). "Possible evolution of splice-junction signals in eukaryotic genes from stop codons". Proceedings of the National Academy of Sciences of the United States of America. 85 (4): 1129–1133. Bibcode:1988PNAS...85.1129S. doi:10.1073/pnas.85.4.1129. ISSN 0027-8424. PMC 279719. PMID 3422483.
- Senapathy, P. (1995-06-02). "Introns and the origin of protein-coding genes". Science. 268 (5215): 1366–1367, author reply 1367–1369. Bibcode:1995Sci...268.1366S. doi:10.1126/science.7761858. ISSN 0036-8075. PMID 7761858.
- Theobald, Douglas L. (2012). "29+ Evidences for Macroevolution: The Scientific Case for Common Descent". Cite journal requires
|journal=(help) - Gillies, S. D.; Morrison, S. L.; Oi, V. T.; Tonegawa, S. (June 1983). "A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene". Cell. 33 (3): 717–728. doi:10.1016/0092-8674(83)90014-4. ISSN 0092-8674. PMID 6409417.
- Mercola, M.; Wang, X. F.; Olsen, J.; Calame, K. (1983-08-12). "Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus". Science. 221 (4611): 663–665. Bibcode:1983Sci...221..663M. doi:10.1126/science.6306772. ISSN 0036-8075. PMID 6306772.
- Berk, A. J.; Sharp, P. A. (November 1977). "Sizing and mapping of early adenovirus mRNAs by gel electrophoresis of S1 endonuclease-digested hybrids". Cell. 12 (3): 721–732. doi:10.1016/0092-8674(77)90272-0. ISSN 0092-8674. PMID 922889.
- Berget, S M; Moore, C; Sharp, P A (August 1977). "Spliced segments at the 5' terminus of adenovirus 2 late mRNA". Proceedings of the National Academy of Sciences of the United States of America. 74 (8): 3171–3175. Bibcode:1977PNAS...74.3171B. doi:10.1073/pnas.74.8.3171. ISSN 0027-8424. PMC 431482. PMID 269380.
- Chow, L. T.; Roberts, J. M.; Lewis, J. B.; Broker, T. R. (August 1977). "A map of cytoplasmic RNA transcripts from lytic adenovirus type 2, determined by electron microscopy of RNA:DNA hybrids". Cell. 11 (4): 819–836. doi:10.1016/0092-8674(77)90294-X. ISSN 0092-8674. PMID 890740.
- "Online Education Kit: 1977: Introns Discovered". National Human Genome Research Institute (NHGRI). Retrieved 2019-01-01.
- Doolittle, W. Ford (13 April 1978). "Genes in pieces: were they ever together?". Nature. 272 (5654): 581–582. Bibcode:1978Natur.272..581D. doi:10.1038/272581a0. ISSN 1476-4687.
- Darnell, J. E. (1978-12-22). "Implications of RNA-RNA splicing in evolution of eukaryotic cells". Science. 202 (4374): 1257–1260. doi:10.1126/science.364651. ISSN 0036-8075. PMID 364651.
- Doolittle, W. F.; Darnell, J. E. (1986-03-01). "Speculations on the early course of evolution". Proceedings of the National Academy of Sciences. 83 (5): 1271–1275. Bibcode:1986PNAS...83.1271D. doi:10.1073/pnas.83.5.1271. ISSN 1091-6490. PMC 323057. PMID 2419905.
- Blake, C.C.F. (1985-01-01). Exons and the Evolution of Proteins. International Review of Cytology. 93. pp. 149–185. doi:10.1016/S0074-7696(08)61374-1. ISBN 9780123644930. ISSN 0074-7696.
- Gilbert, Walter (February 1978). "Why genes in pieces?". Nature. 271 (5645): 501. Bibcode:1978Natur.271..501G. doi:10.1038/271501a0. ISSN 1476-4687. PMID 622185.
- Tonegawa, S; Maxam, A M; Tizard, R; Bernard, O; Gilbert, W (March 1978). "Sequence of a mouse germ-line gene for a variable region of an immunoglobulin light chain". Proceedings of the National Academy of Sciences of the United States of America. 75 (3): 1485–1489. Bibcode:1978PNAS...75.1485T. doi:10.1073/pnas.75.3.1485. ISSN 0027-8424. PMC 411497. PMID 418414.
- Feng, D. F.; Doolittle, R. F. (1987-01-01). "Reconstructing the Evolution of Vertebrate Blood Coagulation from a Consideration of the Amino Acid Sequences of Clotting Proteins". Cold Spring Harbor Symposia on Quantitative Biology. 52: 869–874. doi:10.1101/SQB.1987.052.01.095. ISSN 1943-4456. PMID 3483343.
- Gibbons, A. (1990-12-07). "Calculating the original family--of exons". Science. 250 (4986): 1342. Bibcode:1990Sci...250.1342G. doi:10.1126/science.1701567. ISSN 1095-9203. PMID 1701567.
- Reva, Oleg; Tümmler, Burkhard (2008). "Think big – giant genes in bacteria" (PDF). Environmental Microbiology. 10 (3): 768–777. doi:10.1111/j.1462-2920.2007.01500.x. hdl:2263/9009. ISSN 1462-2920. PMID 18237309.
- Regulapati, Rahul; Singh, Chandan Kumar; Bhasi, Ashwini; Senapathy, Periannan (2008-10-20). "Origination of the Split Structure of Spliceosomal Genes from Random Genetic Sequences". PLOS ONE. 3 (10): e3456. Bibcode:2008PLoSO...3.3456R. doi:10.1371/journal.pone.0003456. ISSN 1932-6203. PMC 2565106. PMID 18941625.
- Information, Reed Business (1986-06-26). New Scientist. Reed Business Information.
- Information, Reed Business (1988-03-31). New Scientist. Reed Business Information.
- Senapathy, Periannan; Harris, Nomi L. (1990-05-25). "Distribution and consenus of branch point signals in eukaryotic genes: a computerized statistical analysis". Nucleic Acids Research. 18 (10): 3015–9. doi:10.1093/nar/18.10.3015. ISSN 0305-1048. PMC 330832. PMID 2349097.
- Maier, U.-G.; Brown, J.W.S.; Toloczyki, C.; Feix, G. (January 1987). "Binding of a nuclear factor to a consensus sequence in the 5' flanking region of zein genes from maize". The EMBO Journal. 6 (1): 17–22. doi:10.1002/j.1460-2075.1987.tb04712.x. ISSN 0261-4189. PMC 553350. PMID 15981330.
- Keller, E B; Noon, W A (1985-07-11). "Intron splicing: a conserved internal signal in introns of Drosophila pre-mRNAs". Nucleic Acids Research. 13 (13): 4971–4981. doi:10.1093/nar/13.13.4971. ISSN 0305-1048. PMC 321838. PMID 2410858.
- BIRNSTIEL, M; BUSSLINGER, M; STRUB, K (June 1985). "Transcription termination and 3′ processing: the end is in site!". Cell. 41 (2): 349–359. doi:10.1016/s0092-8674(85)80007-6. ISSN 0092-8674.
- Consortium, International Human Genome Sequencing (February 2001). "Initial sequencing and analysis of the human genome". Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. ISSN 1476-4687. PMID 11237011.
- Zhu, Xiaohong; Zandieh, Ali; Xia, Ashley; Wu, Mitchell; Wu, David; Wen, Meiyuan; Wang, Mei; Venter, Eli; Turner, Russell (2001-02-16). "The Sequence of the Human Genome". Science. 291 (5507): 1304–1351. Bibcode:2001Sci...291.1304V. doi:10.1126/science.1058040. ISSN 1095-9203. PMID 11181995.
- Kang, Byoung-Cheorl; Nah, Gyoungju; Lee, Heung-Ryul; Han, Koeun; Purushotham, Preethi M.; Jo, Jinkwan (2017). "Development of a Genetic Map for Onion (Allium cepa L.) Using Reference-Free Genotyping-by-Sequencing and SNP Assays". Frontiers in Plant Science. 8: 1606. doi:10.3389/fpls.2017.01606. ISSN 1664-462X. PMC 5604068. PMID 28959273.
- Smith, Jeramiah J.; Voss, S. Randal; Tsonis, Panagiotis A.; Timoshevskaya, Nataliya Y.; Timoshevskiy, Vladimir A.; Keinath, Melissa C. (2015-11-10). "Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing". Scientific Reports. 5: 16413. Bibcode:2015NatSR...516413K. doi:10.1038/srep16413. ISSN 2045-2322. PMC 4639759. PMID 26553646.
- Venter, J. C.; Adams, M. D.; Myers, E. W.; Li, P. W.; Mural, R. J.; Sutton, G. G.; Smith, H. O.; Yandell, M.; Evans, C. A. (2001-02-16). "The sequence of the human genome". Science. 291 (5507): 1304–1351. Bibcode:2001Sci...291.1304V. doi:10.1126/science.1058040. ISSN 0036-8075. PMID 11181995.
- Lander, E. S.; Linton, L. M.; Birren, B.; Nusbaum, C.; Zody, M. C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M. (2001-02-15). "Initial sequencing and analysis of the human genome" (PDF). Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. ISSN 0028-0836. PMID 11237011.
- Consortium*, The C. elegans Sequencing (1998-12-11). "Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology". Science. 282 (5396): 2012–2018. Bibcode:1998Sci...282.2012.. doi:10.1126/science.282.5396.2012. ISSN 1095-9203. PMID 9851916.
- Arabidopsis Genome Initiative (2000-12-14). "Analysis of the genome sequence of the flowering plant Arabidopsis thaliana". Nature. 408 (6814): 796–815. Bibcode:2000Natur.408..796T. doi:10.1038/35048692. ISSN 0028-0836. PMID 11130711.
- Bennetzen, Jeffrey L.; Brown, James K. M.; Devos, Katrien M. (2002-07-01). "Genome Size Reduction through Illegitimate Recombination Counteracts Genome Expansion in Arabidopsis". Genome Research. 12 (7): 1075–1079. doi:10.1101/gr.132102. ISSN 1549-5469. PMC 186626. PMID 12097344.
- Kurland, C. G.; Canbäck, B.; Berg, O. G. (December 2007). "The origins of modern proteomes". Biochimie. 89 (12): 1454–1463. doi:10.1016/j.biochi.2007.09.004. ISSN 0300-9084. PMID 17949885.
- Caetano-Anollés, Gustavo; Caetano-Anollés, Derek (July 2003). "An evolutionarily structured universe of protein architecture". Genome Research. 13 (7): 1563–1571. doi:10.1101/gr.1161903. ISSN 1088-9051. PMC 403752. PMID 12840035.
- Glansdorff, Nicolas; Xu, Ying; Labedan, Bernard (2008-07-09). "The last universal common ancestor: emergence, constitution and genetic legacy of an elusive forerunner". Biology Direct. 3: 29. doi:10.1186/1745-6150-3-29. ISSN 1745-6150. PMC 2478661. PMID 18613974.
- Kurland, C. G.; Collins, L. J.; Penny, D. (2006-05-19). "Genomics and the irreducible nature of eukaryote cells". Science. 312 (5776): 1011–1014. Bibcode:2006Sci...312.1011K. doi:10.1126/science.1121674. ISSN 1095-9203. PMID 16709776.
- Collins, Lesley; Penny, David (April 2005). "Complex spliceosomal organization ancestral to extant eukaryotes". Molecular Biology and Evolution. 22 (4): 1053–1066. doi:10.1093/molbev/msi091. ISSN 0737-4038. PMID 15659557.
- Penny, David; Collins, Lesley J.; Daly, Toni K.; Cox, Simon J. (December 2014). "The relative ages of eukaryotes and akaryotes". Journal of Molecular Evolution. 79 (5–6): 228–239. Bibcode:2014JMolE..79..228P. doi:10.1007/s00239-014-9643-y. ISSN 1432-1432. PMID 25179144.
- Fuerst, John A.; Sagulenko, Evgeny (2012-05-04). "Keys to Eukaryality: Planctomycetes and Ancestral Evolution of Cellular Complexity". Frontiers in Microbiology. 3: 167. doi:10.3389/fmicb.2012.00167. ISSN 1664-302X. PMC 3343278. PMID 22586422.
- Collins, Lesley; Penny, David (April 2005). "Complex spliceosomal organization ancestral to extant eukaryotes". Molecular Biology and Evolution. 22 (4): 1053–1066. doi:10.1093/molbev/msi091. ISSN 0737-4038. PMID 15659557.
- Shapiro, M. B.; Senapathy, P. (1987-09-11). "RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression". Nucleic Acids Research. 15 (17): 7155–7174. doi:10.1093/nar/15.17.7155. ISSN 0305-1048. PMC 306199. PMID 3658675.
- Senapathy, P.; Shapiro, M. B.; Harris, N. L. (1990). Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods in Enzymology. 183. pp. 252–278. doi:10.1016/0076-6879(90)83018-5. ISBN 9780121820848. ISSN 0076-6879. PMID 2314278.
- "National Institutes of Health (NIH) — All of Us". allofus.nih.gov. Retrieved 2019-01-02.
- Penny, David; Collins, Lesley (2005-04-01). "Complex Spliceosomal Organization Ancestral to Extant Eukaryotes". Molecular Biology and Evolution. 22 (4): 1053–1066. doi:10.1093/molbev/msi091. ISSN 0737-4038. PMID 15659557.
- Caetano-Anollés, Derek; Caetano-Anollés, Gustavo (2003-07-01). "An Evolutionarily Structured Universe of Protein Architecture". Genome Research. 13 (7): 1563–1571. doi:10.1101/gr.1161903. ISSN 1549-5469. PMC 403752. PMID 12840035.
- Glansdorff, Nicolas; Xu, Ying; Labedan, Bernard (2008-07-09). "The Last Universal Common Ancestor: emergence, constitution and genetic legacy of an elusive forerunner". Biology Direct. 3 (1): 29. doi:10.1186/1745-6150-3-29. ISSN 1745-6150. PMC 2478661. PMID 18613974.
- Kurland, C.G.; Canbäck, B.; Berg, O.G. (2007-12-01). "The origins of modern proteomes". Biochimie. 89 (12): 1454–1463. doi:10.1016/j.biochi.2007.09.004. ISSN 0300-9084. PMID 17949885.
- Penny, D.; Collins, L. J.; Kurland, C. G. (2006-05-19). "Genomics and the Irreducible Nature of Eukaryote Cells". Science. 312 (5776): 1011–1014. Bibcode:2006Sci...312.1011K. doi:10.1126/science.1121674. ISSN 1095-9203. PMID 16709776.
- Poole, A. M.; Jeffares, D. C.; Penny, D. (January 1998). "The path from the RNA world". Journal of Molecular Evolution. 46 (1): 1–17. Bibcode:1998JMolE..46....1P. doi:10.1007/PL00006275. ISSN 0022-2844. PMID 9419221.
- Forterre, Patrick; Philippe, Hervé (1999). "Where is the root of the universal tree of life?". BioEssays. 21 (10): 871–879. doi:10.1002/(SICI)1521-1878(199910)21:10<871::AID-BIES10>3.0.CO;2-Q. ISSN 1521-1878. PMID 10497338.
- Cox, Simon J.; Daly, Toni K.; Collins, Lesley J.; Penny, David (2014-12-01). "The Relative Ages of Eukaryotes and Akaryotes". Journal of Molecular Evolution. 79 (5–6): 228–239. Bibcode:2014JMolE..79..228P. doi:10.1007/s00239-014-9643-y. ISSN 1432-1432. PMID 25179144.
- Sagulenko, Evgeny; Fuerst, John Arlington (2012). "Keys to eukaryality: planctomycetes and ancestral evolution of cellular complexity". Frontiers in Microbiology. 3. doi:10.3389/fmicb.2012.00167. ISSN 1664-302X. PMC 3343278. PMID 22586422.
- Gilbert, Walter; Roy, Scott W. (2005-02-08). "Complex early genes". Proceedings of the National Academy of Sciences. 102 (6): 1986–1991. Bibcode:2005PNAS..102.1986R. doi:10.1073/pnas.0408355101. ISSN 1091-6490. PMC 548548. PMID 15687506.
- Gilbert, Walter; Roy, Scott William (March 2006). "The evolution of spliceosomal introns: patterns, puzzles and progress". Nature Reviews Genetics. 7 (3): 211–221. doi:10.1038/nrg1807. ISSN 1471-0064. PMID 16485020.
- Rogozin, Igor B.; Sverdlov, Alexander V.; Babenko, Vladimir N.; Koonin, Eugene V. (June 2005). "Analysis of evolution of exon-intron structure of eukaryotic genes". Briefings in Bioinformatics. 6 (2): 118–134. doi:10.1093/bib/6.2.118. ISSN 1467-5463. PMID 15975222.
- Sullivan, James C.; Reitzel, Adam M.; Finnerty, John R. (2006). "A high percentage of introns in human genes were present early in animal evolution: evidence from the basal metazoan Nematostella vectensis". Genome Informatics. International Conference on Genome Informatics. 17 (1): 219–229. ISSN 0919-9454. PMID 17503371.
- Koonin, Eugene V.; Rogozin, Igor B.; Csuros, Miklos (2011-09-15). "A Detailed History of Intron-rich Eukaryotic Ancestors Inferred from a Global Survey of 100 Complete Genomes". PLOS Computational Biology. 7 (9): e1002150. Bibcode:2011PLSCB...7E2150C. doi:10.1371/journal.pcbi.1002150. ISSN 1553-7358. PMC 3174169. PMID 21935348.