Single-nucleotide polymorphism
A single-nucleotide polymorphism (SNP; /snɪp/; plural /snɪps/) is a substitution of a single nucleotide at a specific position in the genome, that is present in a sufficiently large fraction of the population (e.g. 1% or more).[1]
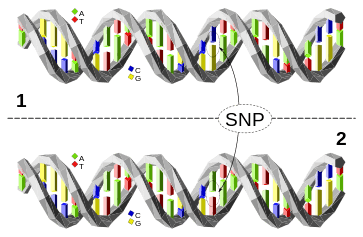
For example, at a specific base position in the human genome, the C nucleotide may appear in most individuals, but in a minority of individuals, the position is occupied by an A. This means that there is a SNP at this specific position, and the two possible nucleotide variations – C or A – are said to be the alleles for this specific position.
SNPs pinpoint differences in our susceptibility to a wide range of diseases (e.g. sickle-cell anemia, β-thalassemia and cystic fibrosis result from SNPs).[2][3][4] The severity of illness and the way the body responds to treatments are also manifestations of genetic variations. For example, a single-base mutation in the APOE (apolipoprotein E) gene is associated with a lower risk for Alzheimer's disease.[5]
A single-nucleotide variant (SNV) is a variation in a single nucleotide without any limitations of frequency and may arise in somatic cells. A somatic single-nucleotide variation (e.g., caused by cancer) may also be called a single-nucleotide alteration.
Types
| Types of SNPs |
|---|
|
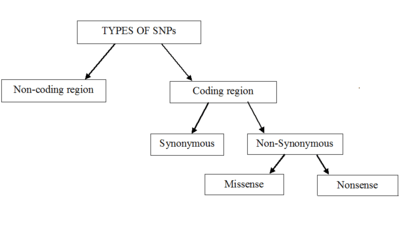
Single-nucleotide polymorphisms may fall within coding sequences of genes, non-coding regions of genes, or in the intergenic regions (regions between genes). SNPs within a coding sequence do not necessarily change the amino acid sequence of the protein that is produced, due to degeneracy of the genetic code.
SNPs in the coding region are of two types: synonymous and nonsynonymous SNPs. Synonymous SNPs do not affect the protein sequence, while nonsynonymous SNPs change the amino acid sequence of protein. The nonsynonymous SNPs are of two types: missense and nonsense.
SNPs that are not in protein-coding regions may still affect gene splicing, transcription factor binding, messenger RNA degradation, or the sequence of noncoding RNA. Gene expression affected by this type of SNP is referred to as an eSNP (expression SNP) and may be upstream or downstream from the gene.
Applications
- Association studies can determine whether a genetic variant is associated with a disease or trait.[6]
- A tag SNP is a representative single-nucleotide polymorphism in a region of the genome with high linkage disequilibrium (the non-random association of alleles at two or more loci). Tag SNPs are useful in whole-genome SNP association studies, in which hundreds of thousands of SNPs across the entire genome are genotyped.
- Haplotype mapping: sets of alleles or DNA sequences can be clustered so that a single SNP can identify many linked SNPs.
- Linkage disequilibrium (LD), a term used in population genetics, indicates non-random association of alleles at two or more loci, not necessarily on the same chromosome. It refers to the phenomenon that SNP allele or DNA sequence that are close together in the genome tend to be inherited together. LD is affected by two parameters: 1) The distance between the SNPs [the larger the distance, the lower the LD]. 2) Recombination rate [the lower the recombination rate, the higher the LD].[7]
Frequency
More than 335 million SNPs have been found across humans from multiple populations. A typical genome differs from the reference human genome at 4 to 5 million sites, most of which (more than 99.9%) consist of SNPs and short indels.[8]
Within a genome
The genomic distribution of SNPs is not homogenous; SNPs occur in non-coding regions more frequently than in coding regions or, in general, where natural selection is acting and "fixing" the allele (eliminating other variants) of the SNP that constitutes the most favorable genetic adaptation.[9] Other factors, like genetic recombination and mutation rate, can also determine SNP density.[10]
SNP density can be predicted by the presence of microsatellites: AT microsatellites in particular are potent predictors of SNP density, with long (AT)(n) repeat tracts tending to be found in regions of significantly reduced SNP density and low GC content.[11]
Within a population
There are variations between human populations, so a SNP allele that is common in one geographical or ethnic group may be much rarer in another. Within a population, SNPs can be assigned a minor allele frequency—the lowest allele frequency at a locus that is observed in a particular population.[12] This is simply the lesser of the two allele frequencies for single-nucleotide polymorphisms.
Importance
Variations in the DNA sequences of humans can affect how humans develop diseases and respond to pathogens, chemicals, drugs, vaccines, and other agents. SNPs are also critical for personalized medicine.[13] Examples include biomedical research, forensics, pharmacogenetics, and disease causation, as outlined below.
Clinical research
SNPs' greatest importance in clinical research is for comparing regions of the genome between cohorts (such as with matched cohorts with and without a disease) in genome-wide association studies. SNPs have been used in genome-wide association studies as high-resolution markers in gene mapping related to diseases or normal traits.[14] SNPs without an observable impact on the phenotype (so called silent mutations) are still useful as genetic markers in genome-wide association studies, because of their quantity and the stable inheritance over generations.[15]
Forensics
SNPs were used initially for matching a forensic DNA sample to a suspect but it has been phased out with development of STR-based DNA fingerprinting techniques.[16] Current next-generation-sequencing (NGS) techniques may allow for better use of SNP genotyping in a forensic application so long as problematic loci are avoided.[17] In the future SNPs may be used in forensics for some phenotypic clues like eye color, hair color, ethnicity, etc. Kidd et al. have demonstrated that a panel of 19 SNPs can identify the ethnic group with good probability of match (Pm = 10−7) in 40 population groups studied.[18] One example of how this might potentially be useful is in the area of artistic reconstruction of possible premortem appearances of skeletal remains of unknown individuals. Although a facial reconstruction can be fairly accurate based strictly upon anthropological features, other data that might allow a more accurate representation include eye color, skin color, hair color, etc.
In a situation with a low amount of forensic sample or a degraded sample, SNP methods can be a good alternative to STR methods due to the abundance of potential markers, amenability to automation, and potential reduction of required fragment length to only 60–80 bp.[19] In the absence of a STR match in DNA profile database; different SNPs can be used to get clues regarding ethnicity, phenotype, lineage, and even identity.
Pharmacogenetics
Some SNPs are associated with the metabolism of different drugs.[20][21][22] SNP's can be mutations, such as deletions, which can inhibit or promote enzymatic activity; such change in enzymatic activity can lead to decreased rates of drug metabolism.[23] The association of a wide range of human diseases like cancer, infectious diseases (AIDS, leprosy, hepatitis, etc.) autoimmune, neuropsychiatric and many other diseases with different SNPs can be made as relevant pharmacogenomic targets for drug therapy.[24]
Disease
A single SNP may cause a Mendelian disease, though for complex diseases, SNPs do not usually function individually, rather, they work in coordination with other SNPs to manifest a disease condition as has been seen in Osteoporosis.[25] One of the earliest successes in this field was finding a single base mutation in the non-coding region of the APOC3 (apolipoprotein C3 gene) that associated with higher risks of hypertriglyceridemia and atherosclerosis.[26]
All types of SNPs can have an observable phenotype or can result in disease:
- SNPs in non-coding regions can manifest in a higher risk of cancer,[27] and may affect mRNA structure and disease susceptibility.[28] Non-coding SNPs can also alter the level of expression of a gene, as an eQTL (expression quantitative trait locus).
- SNPs in coding regions:
- synonymous substitutions by definition do not result in a change of amino acid in the protein, but still can affect its function in other ways. An example would be a seemingly silent mutation in the multidrug resistance gene 1 (MDR1), which codes for a cellular membrane pump that expels drugs from the cell, can slow down translation and allow the peptide chain to fold into an unusual conformation, causing the mutant pump to be less functional (in MDR1 protein e.g. C1236T polymorphism changes a GGC codon to GGT at amino acid position 412 of the polypeptide (both encode glycine) and the C3435T polymorphism changes ATC to ATT at position 1145 (both encode isoleucine)).[29]
- nonsynonymous substitutions:
- missense – single change in the base results in change in amino acid of protein and its malfunction which leads to disease (e.g. c.1580G>T SNP in LMNA gene – position 1580 (nt) in the DNA sequence (CGT codon) causing the guanine to be replaced with the thymine, yielding CTT codon in the DNA sequence, results at the protein level in the replacement of the arginine by the leucine in the position 527,[30] at the phenotype level this manifests in overlapping mandibuloacral dysplasia and progeria syndrome)
- nonsense – point mutation in a sequence of DNA that results in a premature stop codon, or a nonsense codon in the transcribed mRNA, and in a truncated, incomplete, and usually nonfunctional protein product (e.g. Cystic fibrosis caused by the G542X mutation in the cystic fibrosis transmembrane conductance regulator gene).[31]
Examples
- rs6311 and rs6313 are SNPs in the Serotonin 5-HT2A receptor gene on human chromosome 13.[32]
- A SNP in the F5 gene causes Factor V Leiden thrombophilia.[33]
- rs3091244 is an example of a triallelic SNP in the CRP gene on human chromosome 1.[34]
- TAS2R38 codes for PTC tasting ability, and contains 6 annotated SNPs.[35]
- rs148649884 and rs138055828 in the FCN1 gene encoding M-ficolin crippled the ligand-binding capability of the recombinant M-ficolin.[36]
- An intronic SNP in DNA mismatch repair gene PMS2 (rs1059060, Ser775Asn) is associated with increased sperm DNA damage and risk of male infertility.[37]
Databases
As there are for genes, bioinformatics databases exist for SNPs.
- dbSNP is a SNP database from the National Center for Biotechnology Information (NCBI). As of June 8, 2015, dbSNP listed 149,735,377 SNPs in humans.[38][39]
- Kaviar[40] is a compendium of SNPs from multiple data sources including dbSNP.
- SNPedia is a wiki-style database supporting personal genome annotation, interpretation and analysis.
- The OMIM database describes the association between polymorphisms and diseases (e.g., gives diseases in text form)
- dbSAP – single amino-acid polymorphism database for protein variation detection[41]
- The Human Gene Mutation Database provides gene mutations causing or associated with human inherited diseases and functional SNPs
- The International HapMap Project, where researchers are identifying Tag SNPs to be able to determine the collection of haplotypes present in each subject.
- GWAS Central allows users to visually interrogate the actual summary-level association data in one or more genome-wide association studies.
The International SNP Map working group mapped the sequence flanking each SNP by alignment to the genomic sequence of large-insert clones in Genebank. These alignments were converted to chromosomal coordinates that is shown in Table 1.[42] This list has greatly increased since, with, for instance, the Kaviar database now listing 162 million single nucleotide variants (SNVs).
| Chromosome | Length(bp) | All SNPs | TSC SNPs | ||
|---|---|---|---|---|---|
| Total SNPs | kb per SNP | Total SNPs | kb per SNP | ||
| 1 | 214,066,000 | 129,931 | 1.65 | 75,166 | 2.85 |
| 2 | 222,889,000 | 103,664 | 2.15 | 76,985 | 2.90 |
| 3 | 186,938,000 | 93,140 | 2.01 | 63,669 | 2.94 |
| 4 | 169,035,000 | 84,426 | 2.00 | 65,719 | 2.57 |
| 5 | 170,954,000 | 117,882 | 1.45 | 63,545 | 2.69 |
| 6 | 165,022,000 | 96,317 | 1.71 | 53,797 | 3.07 |
| 7 | 149,414,000 | 71,752 | 2.08 | 42,327 | 3.53 |
| 8 | 125,148,000 | 57,834 | 2.16 | 42,653 | 2.93 |
| 9 | 107,440,000 | 62,013 | 1.73 | 43,020 | 2.50 |
| 10 | 127,894,000 | 61,298 | 2.09 | 42,466 | 3.01 |
| 11 | 129,193,000 | 84,663 | 1.53 | 47,621 | 2.71 |
| 12 | 125,198,000 | 59,245 | 2.11 | 38,136 | 3.28 |
| 13 | 93,711,000 | 53,093 | 1.77 | 35,745 | 2.62 |
| 14 | 89,344,000 | 44,112 | 2.03 | 29,746 | 3.00 |
| 15 | 73,467,000 | 37,814 | 1.94 | 26,524 | 2.77 |
| 16 | 74,037,000 | 38,735 | 1.91 | 23,328 | 3.17 |
| 17 | 73,367,000 | 34,621 | 2.12 | 19,396 | 3.78 |
| 18 | 73,078,000 | 45,135 | 1.62 | 27,028 | 2.70 |
| 19 | 56,044,000 | 25,676 | 2.18 | 11,185 | 5.01 |
| 20 | 63,317,000 | 29,478 | 2.15 | 17,051 | 3.71 |
| 21 | 33,824,000 | 20,916 | 1.62 | 9,103 | 3.72 |
| 22 | 33,786,000 | 28,410 | 1.19 | 11,056 | 3.06 |
| X | 131,245,000 | 34,842 | 3.77 | 20,400 | 6.43 |
| Y | 21,753,000 | 4,193 | 5.19 | 1,784 | 12.19 |
| RefSeq | 15,696,674 | 14,534 | 1.08 | ||
| Totals | 2,710,164,000 | 1,419,190 | 1.91 | 887,450 | 3.05 |
Nomenclature
The nomenclature for SNPs can be confusing: several variations can exist for an individual SNP, and consensus has not yet been achieved.
The rs### standard is that which has been adopted by dbSNP and uses the prefix "rs", for "reference SNP", followed by a unique and arbitrary number.[43] SNPs are frequently referred to by their dbSNP rs number, as in the examples above.
The Human Genome Variation Society (HGVS) uses a standard which conveys more information about the SNP. Examples are:
- c.76A>T: "c." for coding region, followed by a number for the position of the nucleotide, followed by a one-letter abbreviation for the nucleotide (A, C, G, T or U), followed by a greater than sign (">") to indicate substitution, followed by the abbreviation of the nucleotide which replaces the former[44][45][46]
- p.Ser123Arg: "p." for protein, followed by a three-letter abbreviation for the amino acid, followed by a number for the position of the amino acid, followed by the abbreviation of the amino acid which replaces the former.[47]
SNP analysis
SNPs are usually biallelic and thus easily assayed.[48] Analytical methods to discover novel SNPs and detect known SNPs include:
- DNA sequencing;[49]
- capillary electrophoresis;[50]
- mass spectrometry;[51]
- single-strand conformation polymorphism (SSCP);[52]
- single-base extension;
- electrochemical analysis;
- denaturating HPLC and gel electrophoresis;
- restriction fragment length polymorphism;
- hybridization analysis;
Programs for prediction of SNP effects
An important group of SNPs are those that corresponds to missense mutations causing amino acid change on protein level. Point mutation of particular residue can have different effect on protein function (from no effect to complete disruption its function). Usually, change in amino acids with similar size and physico-chemical properties (e.g. substitution from leucine to valine) has mild effect, and opposite. Similarly, if SNP disrupts secondary structure elements (e.g. substitution to proline in alpha helix region) such mutation usually may affect whole protein structure and function. Using those simple and many other machine learning derived rules a group of programs for the prediction of SNP effect was developed:
- SIFT This program provides insight into how a laboratory induced missense or nonsynonymous mutation will affect protein function based on physical properties of the amino acid and sequence homology.
- LIST[53][54] (Local Identity and Shared Taxa) estimates the potential deleteriousness of mutations resulted from altering their protein functions. It is based on the assumption that variations observed in closely related species are more significant when assessing conservation compared to those in distantly related species.
- SNAP2
- SuSPect
- PolyPhen-2
- PredictSNP
- MutationTaster: official website
- Variant Effect Predictor from the Ensembl project
- SNPViz[55] This program provides a 3D representation of the protein affected, highlighting the amino acid change so doctors can determine pathogenicity of the mutant protein.
- PROVEAN
- PhyreRisk is a database which maps variants to experimental and predicted protein structures.[56]
- Missense3D is a tool which provides a stereochemical report on the effect of missense variants on protein structure.[57]
See also
- Affymetrix
- HapMap
- Illumina
- International HapMap Project
- Short tandem repeat (STR)
- Single-base extension
- SNP array
- SNP genotyping
- SNPedia
- Snpstr
- SNV calling from NGS data
- Tag SNP
- TaqMan
- Variome
References
- "single-nucleotide polymorphism / SNP | Learn Science at Scitable". www.nature.com. Archived from the original on 2015-11-10. Retrieved 2015-11-13.
- Ingram VM (October 1956). "A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin". Nature. 178 (4537): 792–4. Bibcode:1956Natur.178..792I. doi:10.1038/178792a0. PMID 13369537. S2CID 4167855.
- Chang JC, Kan YW (June 1979). "beta 0 thalassemia, a nonsense mutation in man". Proceedings of the National Academy of Sciences of the United States of America. 76 (6): 2886–9. Bibcode:1979PNAS...76.2886C. doi:10.1073/pnas.76.6.2886. PMC 383714. PMID 88735.
- Hamosh A, King TM, Rosenstein BJ, Corey M, Levison H, Durie P, Tsui LC, McIntosh I, Keston M, Brock DJ (August 1992). "Cystic fibrosis patients bearing both the common missense mutation Gly----Asp at codon 551 and the delta F508 mutation are clinically indistinguishable from delta F508 homozygotes, except for decreased risk of meconium ileus". American Journal of Human Genetics. 51 (2): 245–50. PMC 1682672. PMID 1379413.
- Wolf AB, Caselli RJ, Reiman EM, Valla J (April 2013). "APOE and neuroenergetics: an emerging paradigm in Alzheimer's disease". Neurobiology of Aging. 34 (4): 1007–17. doi:10.1016/j.neurobiolaging.2012.10.011. PMC 3545040. PMID 23159550.
- Zhang K, Qin ZS, Liu JS, Chen T, Waterman MS, Sun F (May 2004). "Haplotype block partitioning and tag SNP selection using genotype data and their applications to association studies". Genome Research. 14 (5): 908–16. doi:10.1101/gr.1837404. PMC 479119. PMID 15078859.
- Gupta PK, Roy JK, Prasad M (25 February 2001). "Single nucleotide polymorphisms: a new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants". Current Science. 80 (4): 524–535. Archived from the original on 13 February 2017.
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR (October 2015). "A global reference for human genetic variation". Nature. 526 (7571): 68–74. Bibcode:2015Natur.526...68T. doi:10.1038/nature15393. PMC 4750478. PMID 26432245.
- Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L (March 2008). "Natural selection has driven population differentiation in modern humans". Nature Genetics. 40 (3): 340–5. doi:10.1038/ng.78. PMID 18246066. S2CID 205357396.
- Nachman MW (September 2001). "Single nucleotide polymorphisms and recombination rate in humans". Trends in Genetics. 17 (9): 481–5. doi:10.1016/S0168-9525(01)02409-X. PMID 11525814.
- Varela MA, Amos W (March 2010). "Heterogeneous distribution of SNPs in the human genome: microsatellites as predictors of nucleotide diversity and divergence". Genomics. 95 (3): 151–9. doi:10.1016/j.ygeno.2009.12.003. PMID 20026267.
- Zhu Z, Yuan D, Luo D, Lu X, Huang S (2015-07-24). "Enrichment of Minor Alleles of Common SNPs and Improved Risk Prediction for Parkinson's Disease". PLOS ONE. 10 (7): e0133421. Bibcode:2015PLoSO..1033421Z. doi:10.1371/journal.pone.0133421. PMC 4514478. PMID 26207627.
- Carlson, Bruce (15 June 2008). "SNPs — A Shortcut to Personalized Medicine". Genetic Engineering & Biotechnology News. Mary Ann Liebert, Inc. 28 (12). Archived from the original on 26 December 2010. Retrieved 2008-07-06.
(subtitle) Medical applications are where the market's growth is expected
- Yu A, Li F, Xu W, Wang Z, Sun C, Han B, et al. (August 2019). "Application of a high-resolution genetic map for chromosome-scale genome assembly and fine QTLs mapping of seed size and weight traits in castor bean". Scientific Reports. 9 (1): 11950. Bibcode:2019NatSR...911950Y. doi:10.1038/s41598-019-48492-8. PMC 6697702. PMID 31420567.
- Thomas PE, Klinger R, Furlong LI, Hofmann-Apitius M, Friedrich CM (2011). "Challenges in the association of human single nucleotide polymorphism mentions with unique database identifiers". BMC Bioinformatics. 12 Suppl 4: S4. doi:10.1186/1471-2105-12-S4-S4. PMC 3194196. PMID 21992066.
- Butler, John M. (2010). Fundamentals of forensic DNA typing. Burlington, MA: Elsevier/Academic Press. ISBN 9780080961767.
- Cornelis S, Gansemans Y, Deleye L, Deforce D, Van Nieuwerburgh F (February 2017). "Forensic SNP Genotyping using Nanopore MinION Sequencing". Scientific Reports. 7: 41759. Bibcode:2017NatSR...741759C. doi:10.1038/srep41759. PMC 5290523. PMID 28155888.
- Kidd KK, Pakstis AJ, Speed WC, Grigorenko EL, Kajuna SL, Karoma NJ, Kungulilo S, Kim JJ, Lu RB, Odunsi A, Okonofua F, Parnas J, Schulz LO, Zhukova OV, Kidd JR (December 2006). "Developing a SNP panel for forensic identification of individuals". Forensic Science International. 164 (1): 20–32. doi:10.1016/j.forsciint.2005.11.017. PMID 16360294.
- Budowle B, van Daal A (April 2008). "Forensically relevant SNP classes". BioTechniques. 44 (5): 603–8, 610. doi:10.2144/000112806. PMID 18474034.
- Goldstein JA (October 2001). "Clinical relevance of genetic polymorphisms in the human CYP2C subfamily". British Journal of Clinical Pharmacology. 52 (4): 349–55. doi:10.1046/j.0306-5251.2001.01499.x. PMC 2014584. PMID 11678778.
- Lee CR (July–August 2004). "CYP2C9 genotype as a predictor of drug disposition in humans". Methods and Findings in Experimental and Clinical Pharmacology. 26 (6): 463–72. PMID 15349140.
- Yanase K, Tsukahara S, Mitsuhashi J, Sugimoto Y (March 2006). "Functional SNPs of the breast cancer resistance protein-therapeutic effects and inhibitor development". Cancer Letters. 234 (1): 73–80. doi:10.1016/j.canlet.2005.04.039. PMID 16303243.
- Butler, Merlin G. (2018). "Pharmacogenetics and Psychiatric Care: A Review and Commentary". Journal of Mental Health & Clinical Psychology. 2 (2): 17–24. doi:10.29245/2578-2959/2018/2.1120. PMC 6291002. PMID 30556062.
- Fareed M, Afzal M (April 2013). "Single-nucleotide polymorphism in genome-wide association of human population: A tool for broad spectrum service". Egyptian Journal of Medical Human Genetics. 14 (2): 123–134. doi:10.1016/j.ejmhg.2012.08.001.
- Singh M, Singh P, Juneja PK, Singh S, Kaur T (March 2011). "SNP-SNP interactions within APOE gene influence plasma lipids in postmenopausal osteoporosis". Rheumatology International. 31 (3): 421–3. doi:10.1007/s00296-010-1449-7. PMID 20340021. S2CID 32788817.
- Rees A, Shoulders CC, Stocks J, Galton DJ, Baralle FE (February 1983). "DNA polymorphism adjacent to human apoprotein A-1 gene: relation to hypertriglyceridaemia". Lancet. 1 (8322): 444–6. doi:10.1016/S0140-6736(83)91440-X. PMID 6131168. S2CID 29511911.
- Li G, Pan T, Guo D, Li LC (2014). "Regulatory Variants and Disease: The E-Cadherin -160C/A SNP as an Example". Molecular Biology International. 2014: 967565. doi:10.1155/2014/967565. PMC 4167656. PMID 25276428.
- Lu YF, Mauger DM, Goldstein DB, Urban TJ, Weeks KM, Bradrick SS (November 2015). "IFNL3 mRNA structure is remodeled by a functional non-coding polymorphism associated with hepatitis C virus clearance". Scientific Reports. 5: 16037. Bibcode:2015NatSR...516037L. doi:10.1038/srep16037. PMC 4631997. PMID 26531896.
- Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM (January 2007). "A "silent" polymorphism in the MDR1 gene changes substrate specificity". Science. 315 (5811): 525–8. Bibcode:2007Sci...315..525K. doi:10.1126/science.1135308. PMID 17185560.
- Al-Haggar M, Madej-Pilarczyk A, Kozlowski L, Bujnicki JM, Yahia S, Abdel-Hadi D, Shams A, Ahmad N, Hamed S, Puzianowska-Kuznicka M (November 2012). "A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome". European Journal of Human Genetics. 20 (11): 1134–40. doi:10.1038/ejhg.2012.77. PMC 3476705. PMID 22549407.
- Cordovado SK, Hendrix M, Greene CN, Mochal S, Earley MC, Farrell PM, Kharrazi M, Hannon WH, Mueller PW (February 2012). "CFTR mutation analysis and haplotype associations in CF patients". Molecular Genetics and Metabolism. 105 (2): 249–54. doi:10.1016/j.ymgme.2011.10.013. PMC 3551260. PMID 22137130.
- Giegling I, Hartmann AM, Möller HJ, Rujescu D (November 2006). "Anger- and aggression-related traits are associated with polymorphisms in the 5-HT-2A gene". Journal of Affective Disorders. 96 (1–2): 75–81. doi:10.1016/j.jad.2006.05.016. PMID 16814396.
- Kujovich JL (January 2011). "Factor V Leiden thrombophilia". Genetics in Medicine. 13 (1): 1–16. doi:10.1097/GIM.0b013e3181faa0f2. PMID 21116184.
- Morita A, Nakayama T, Doba N, Hinohara S, Mizutani T, Soma M (June 2007). "Genotyping of triallelic SNPs using TaqMan PCR". Molecular and Cellular Probes. 21 (3): 171–6. doi:10.1016/j.mcp.2006.10.005. PMID 17161935.
- Prodi DA, Drayna D, Forabosco P, Palmas MA, Maestrale GB, Piras D, Pirastu M, Angius A (October 2004). "Bitter taste study in a sardinian genetic isolate supports the association of phenylthiocarbamide sensitivity to the TAS2R38 bitter receptor gene". Chemical Senses. 29 (8): 697–702. doi:10.1093/chemse/bjh074. PMID 15466815.
- Ammitzbøll CG, Kjær TR, Steffensen R, Stengaard-Pedersen K, Nielsen HJ, Thiel S, Bøgsted M, Jensenius JC (28 November 2012). "Non-synonymous polymorphisms in the FCN1 gene determine ligand-binding ability and serum levels of M-ficolin". PLOS ONE. 7 (11): e50585. Bibcode:2012PLoSO...750585A. doi:10.1371/journal.pone.0050585. PMC 3509001. PMID 23209787.
- Ji G, Long Y, Zhou Y, Huang C, Gu A, Wang X (May 2012). "Common variants in mismatch repair genes associated with increased risk of sperm DNA damage and male infertility". BMC Medicine. 10: 49. doi:10.1186/1741-7015-10-49. PMC 3378460. PMID 22594646.
- National Center for Biotechnology Information, United States National Library of Medicine. 2014. NCBI dbSNP build 142 for human. "[DBSNP-announce] DBSNP Human Build 142 (GRCh38 and GRCh37.p13)". Archived from the original on 2017-09-10. Retrieved 2017-09-11.
- National Center for Biotechnology Information, United States National Library of Medicine. 2015. NCBI dbSNP build 144 for human. Summary Page. "DBSNP Summary". Archived from the original on 2017-09-10. Retrieved 2017-09-11.
- Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC (November 2011). "Kaviar: an accessible system for testing SNV novelty". Bioinformatics. 27 (22): 3216–7. doi:10.1093/bioinformatics/btr540. PMC 3208392. PMID 21965822.
- Cao R, Shi Y, Chen S, Ma Y, Chen J, Yang J, Chen G, Shi T (January 2017). "dbSAP: single amino-acid polymorphism database for protein variation detection". Nucleic Acids Research. 45 (D1): D827–D832. doi:10.1093/nar/gkw1096. PMC 5210569. PMID 27903894.
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, Hunt SE, Cole CG, Coggill PC, Rice CM, Ning Z, Rogers J, Bentley DR, Kwok PY, Mardis ER, Yeh RT, Schultz B, Cook L, Davenport R, Dante M, Fulton L, Hillier L, Waterston RH, McPherson JD, Gilman B, Schaffner S, Van Etten WJ, Reich D, Higgins J, Daly MJ, Blumenstiel B, Baldwin J, Stange-Thomann N, Zody MC, Linton L, Lander ES, Altshuler D (February 2001). "A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms". Nature. 409 (6822): 928–33. Bibcode:2001Natur.409..928S. doi:10.1038/35057149. PMID 11237013.
- "Clustered RefSNPs (rs) and Other Data Computed in House". SNP FAQ Archive. Bethesda (MD): U.S. National Center for Biotechnology Information. 2005.
- J.T. Den Dunnen (2008-02-20). "Recommendations for the description of sequence variants". Human Genome Variation Society. Archived from the original on 2008-09-14. Retrieved 2008-09-05.
- den Dunnen JT, Antonarakis SE (2000). "Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion". Human Mutation. 15 (1): 7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. PMID 10612815.
- Ogino S, Gulley ML, den Dunnen JT, Wilson RB (February 2007). "Standard mutation nomenclature in molecular diagnostics: practical and educational challenges". The Journal of Molecular Diagnostics. 9 (1): 1–6. doi:10.2353/jmoldx.2007.060081. PMC 1867422. PMID 17251329.
- "Sequence Variant Nomenclature". varnomen.hgvs.org. Retrieved 2019-12-02.
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, Hunt SE, Cole CG, Coggill PC, Rice CM, Ning Z, Rogers J, Bentley DR, Kwok PY, Mardis ER, Yeh RT, Schultz B, Cook L, Davenport R, Dante M, Fulton L, Hillier L, Waterston RH, McPherson JD, Gilman B, Schaffner S, Van Etten WJ, Reich D, Higgins J, Daly MJ, Blumenstiel B, Baldwin J, Stange-Thomann N, Zody MC, Linton L, Lander ES, Altshuler D (February 2001). "A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms". Nature. 409 (6822): 928–33. Bibcode:2001Natur.409..928S. doi:10.1038/35057149. PMID 11237013.
- Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES (September 2000). "An SNP map of the human genome generated by reduced representation shotgun sequencing". Nature. 407 (6803): 513–6. Bibcode:2000Natur.407..513A. doi:10.1038/35035083. PMID 11029002. S2CID 2066435.
- Drabovich AP, Krylov SN (March 2006). "Identification of base pairs in single-nucleotide polymorphisms by MutS protein-mediated capillary electrophoresis". Analytical Chemistry. 78 (6): 2035–8. doi:10.1021/ac0520386. PMID 16536443.
- Griffin TJ, Smith LM (July 2000). "Genetic identification by mass spectrometric analysis of single-nucleotide polymorphisms: ternary encoding of genotypes". Analytical Chemistry. 72 (14): 3298–302. doi:10.1021/ac991390e. PMID 10939403.
- Tahira T, Kukita Y, Higasa K, Okazaki Y, Yoshinaga A, Hayashi K (2009). "Estimation of SNP allele frequencies by SSCP analysis of pooled DNA". Single Nucleotide Polymorphisms. Methods in Molecular Biology. 578. pp. 193–207. doi:10.1007/978-1-60327-411-1_12. ISBN 978-1-60327-410-4. PMID 19768595.
- Malhis N, Jones SJ, Gsponer J (April 2019). "Improved measures for evolutionary conservation that exploit taxonomy distances". Nature Communications. 10 (1): 1556. Bibcode:2019NatCo..10.1556M. doi:10.1038/s41467-019-09583-2. PMC 6450959. PMID 30952844.
- Nawar Malhis; Matthew Jacobson; Steven J. M. Jones; Jörg Gsponer (2020). "LIST-S2: Taxonomy Based Sorting of Deleterious Missense Mutations Across Species". Nucleic Acids Research. doi:10.1093/nar/gkaa288. PMID 32352516.
- "View of SNPViz - Visualization of SNPs in proteins". genomicscomputbiol.org. Retrieved 2018-10-20.
- Ofoegbu TC, David A, Kelley LA, Mezulis S, Islam SA, Mersmann SF, et al. (June 2019). "PhyreRisk: A Dynamic Web Application to Bridge Genomics, Proteomics and 3D Structural Data to Guide Interpretation of Human Genetic Variants". Journal of Molecular Biology. 431 (13): 2460–2466. doi:10.1016/j.jmb.2019.04.043. PMC 6597944. PMID 31075275.
- Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, Sternberg MJ (May 2019). "Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated?". Journal of Molecular Biology. 431 (11): 2197–2212. doi:10.1016/j.jmb.2019.04.009. PMC 6544567. PMID 30995449.
Further reading
- "Glossary". Nature Reviews.
- Human Genome Project Information — SNP Fact Sheet
External links
| Wikimedia Commons has media related to Single nucleotide polymorphism. |
- NCBI resources – Introduction to SNPs from NCBI
- The SNP Consortium LTD – SNP search
- NCBI dbSNP database – "a central repository for both single base nucleotide substitutions and short deletion and insertion polymorphisms"
- HGMD – the Human Gene Mutation Database, includes rare mutations and functional SNPs
- GWAS Central – a central database of summary-level genetic association findings
- 1000 Genomes Project – A Deep Catalog of Human Genetic Variation
- WatCut – an online tool for the design of SNP-RFLP assays
- SNPStats – SNPStats, a web tool for analysis of genetic association studies
- Restriction HomePage – a set of tools for DNA restriction and SNP detection, including design of mutagenic primers
- American Association for Cancer Research Cancer Concepts Factsheet on SNPs
- PharmGKB – The Pharmacogenetics and Pharmacogenomics Knowledge Base, a resource for SNPs associated with drug response and disease outcomes.
- GEN-SNiP – Online tool that identifies polymorphisms in test DNA sequences.
- Rules for Nomenclature of Genes, Genetic Markers, Alleles, and Mutations in Mouse and Rat
- HGNC Guidelines for Human Gene Nomenclature
- SNP effect predictor with galaxy integration
- Open SNP – a portal for sharing own SNP test results
- dbSAP – SNP database for protein variation detection
| Key components | |
|---|---|
| Fields | |
| Archaeogenetics of | |
| Related topics | |
| Lists | |
| |