Delitto perfetto
Delitto perfetto (Italian: [deˈlitto perˈfɛtto]) is a genetic technique for in vivo site-directed mutagenesis in yeast. This name is the Italian term for "perfect murder", and it refers to the ability of the technique to create desired genetic changes without leaving any foreign DNA in the genome.
Background
This technique was developed by a group at the National Institute of Environmental Health Sciences (NIEHS) composed of Michael A. Resnick, Francesca Storici (now at Georgia Institute of Technology), and L. Kevin Lewis (now at Southwest Texas State University). The method uses synthetic oligonucleotides in combination with the cellular process of homologous recombination. Consequently, it is well suited for genetic manipulation of yeast, which has highly efficient homologous recombination. The delitto perfetto approach has been used to produce single and multiple point mutations, gene truncations or insertions, and whole gene deletions (including essential genes).
Advantages
The primary advantage of this technique is its ability to eliminate any foreign DNA from the genome after the mutagenesis process. This ensures there are no selectable markers or exogenous sequences used for targeting left in the genome that may cause unforeseen effects.
The delitto perfetto technique is also simpler compared to other methods for in vivo site-directed mutagenesis. Other methods require multiple cloning steps and extensive DNA sequencing to confirm mutagenesis, which is often a complicated and inefficient process.[1][2][3]
There is great flexibility in this approach because after the CORE cassette is inserted (see Method Overview for details), multiple mutations in the gene of interest can be made easily and quickly.
This method can be applied to other organisms where homologous recombination is efficient, such as the moss Physcomitrella patens, DT40 chicken cells, or E. coli. In addition, human genes can be studied and similarly genetically manipulated in yeast by using yeast artificial chromosomes (YACs).
Disadvantages
Since the delitto perfetto technique is based on homologous recombination, this process must be functional in the cells for the technique to work. In Saccharomyces cerevisiae, the RAD52 gene is essential for homologous recombination, and thus is required for the delitto perfetto method.
The method is useful only for applications where selectable markers are not necessary. For example, mutagenized yeast strains cannot be used for further genetic analysis such as tetrad analysis. Markers would have to be inserted into the appropriate locus in a separate process.
Technical drawbacks
- Cost of oligonucleotides
- Mutagenesis is limited to region of genome surrounding the inserted cassette
- Limited number of reporter genes (restricted by available CORE cassettes)
- Low efficiency for certain applications (e.g. deleting essential genes)
Method overview
Delitto Perfetto is a two step method for in vivo mutagenesis. In the initial step, the CORE cassette is inserted in the region of interest by homologous recombination. Subsequently, the CORE cassette is replaced with DNA containing the mutation of interest.
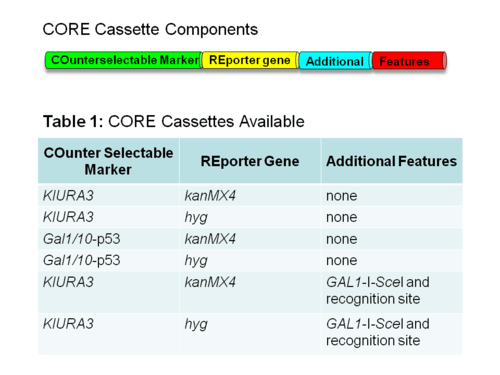
CORE cassettes
The CORE cassette contains both a COunterselectable marker and REporter gene. The reporter gene allows for the selection of yeast cells that receive the CORE cassette during the first step of the process. The counterselectable marker allows for the selection of yeast cells that lose the CORE cassette by the integration of the mutated oligonucleotide during the second step of the process.
There are a variety of CORE cassettes to choose from, which contain a variety of reporter genes, counterselectable makers and additional features.[4]
Reporter genes
- kanMX4 – allows for growth in media containing Geneticin
- hyg – allows for growth in media containing Hygromycin B.
Counterselectable markers
Additional features
- GAL1-I-SceI – Increases the efficiency of targeting to the CORE cassette-containing chromosome in diploid cells. It contains the restriction endonuclease SceI under the GAL1 promoter and the SceI target sequence.[6][7]
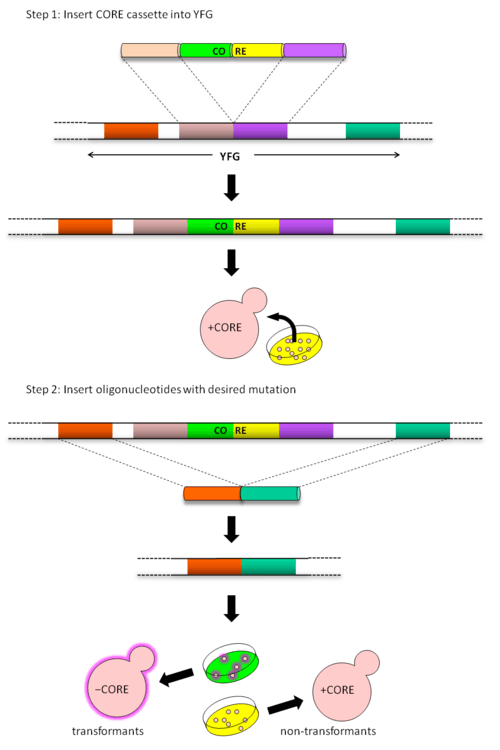
Technique workflow
First the CORE cassette is amplified by PCR with primers containing regions of homology to the chromosomal site where it will be inserted. The CORE cassette is integrated via homologous recombination. Cells containing the CORE cassette can be selected for using the reporter gene and can be further confirmed using the counterselectable marker. Integration of the CORE cassette in the correct chromosomal location can be verified via PCR using primers that anneal upstream of the integration site, within the CORE and downstream of the integration size, which are designed to generate 500–1500 bp fragments.[4]
CORE-containing yeast cells are transformed with oligonucleotides containing the desired mutation such that they lead to the loss of the CORE cassette during homologous recombination. Transformants are selected using the counterselectable marker and can be further screened using the reporter gene. Sequencing is used to ensure the correct mutation has been generated without additional mutations. Alternatively, if the mutation leads to the generation or loss of a restriction site, PCR followed by restriction digest can be used to confirm that the desired mutation has been integrated.[4]
Double-strand break (DSB) mediated delitto perfetto
To increase oligonucleotide targeting to the CORE cassette, CORE cassettes containing the GAL1-I-SceI feature can be used. This feature allows for the expression of SceI, an endonuclease that recognizes a highly unique 18 nucleotide sequence unlikely to occur anywhere else in the S. cerevisiae genome. The SceI endonuclease is able to generate a DSB at the SceI site leading to the recruitment of the DNA repair machinery.[8] This increases the frequency of targeted homologous recombination by 4,000 fold compared to when no DSB is generated.[6]
General considerations for oligonucleotide design
80–100 bp oligonucleotides can be generated as single molecules, or pairs of oligonucleotides that are completely overlapping or partially overlapping. The type of oligonucleotide recommended depends on the type of mutation and the distance from the CORE cassette integration site a mutation is desired.
Longer oligonucleotides lead to increased transformation efficiency. Fully complementary oligonucleotides pairs lead to 5–10 fold increase in efficiency compared to single oligonucleotides and are recommended for all applications.[4][9] However, they provide a small window of mutagenesis of only 20–40 bases from the CORE cassette. To increase the window of mutagenesis, oligonucleotide pairs with a 20 bp overlap can be used, and these allow up to 100 bp upstream and downstream of the CORE integration site to be targeted. However, they transform approximately 6 times less efficiently. To increase the efficiency, partly overlapping oligonucleotides can be extended in vitro.[9]
For gene deletions
Oligonucleotides containing the sequence upstream immediately followed by the sequence downstream of the region to be knocked out are designed. For gene deletions, pairs of fully overlapping 80–100 bp oligonucleotides lead to 5–10 fold increase in transformation efficiency than single oligonucleotides.[4]
For point mutations
For mutations 20 to 40 bp from the CORE cassette, 80–100 bp fully overlapping oligonucleotides are recommended. For mutations more than 40 bp from the CORE cassette, partly overlapping oligonucleotides must be used. To increase their transformation efficiency, it is recommended that partly overlapping oligonucleotides be extended in vitro.[4][6][9]
For essential genes
To generate mutants of essential genes, the CORE cassette can be inserted downstream of the gene of interest, however this limits the regions of the gene available for mutation. Alternatively, diploid cells can be used. However, using a diploid decreases the efficiency of oligonucleotide targeting due to the presence of two suitable chromosomal locations for the oligonucleotides to recombine. To address this drawback, the DSB-mediated delitto perfetto method can be used. This increases the frequency of targeted homologous recombination by 700 fold compared to when no DSB is generated. Moreover, it is 2–5 fold more efficient than other available methods.[6][9]
Background mutation rates
It is reported that when no double stranded breaks are generated, the number of cells that lose the CORE cassette in the absence of a targeting oligonucleotide is less than one transformant per 107 viable cells. In contrast, this number increases up to 100 transformants per 107 viable cells when a double stranded break is generated. In a diploid, the increased background mutation rates occur due to homologous recombination with the homologous chromosome decreasing targeted transformation events to only 4% of the total transformants.[6]
References
- Erdeniz N., Mortensen UH., Rothstein R. (1997) Cloning-free PCR-based allele replacement methods. Genome Res. 7:1174-83.
- Langle-Rouault F., Jacobs E. (1995) A method for performing precise alterations in the yeast genome using a recyclable selectable marker. Nucleic Acids Res. 23:3079-81.
- Scherer S., Davis RW. (1979) Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci. 76:4951-5.
- Storici F., Resnick MA. (2006) The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 409:329-45.
- Inga A., Resnick MA. (2001) Novel human p53 mutations that are toxic to yeast can enhance transactivation of specific promoters and reactive tumor p53 mutants. Oncogene. 14;20(27):3573-9
- Storici F., Durham CL., Gordenin DA., Resnick MA. (2003) Chromosomal site-specific double-strand breaks are efficiently targeted for repair by oligonucleotides in yeast. Proc Natl Acad Sci. 9;100(25):14994-9
- Plessis A., Perrin A., Haber JE., Dujon B. (1992) Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics. 130(3):451-60
- Storici F., Resnick MA. (2003) Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng. 25:189-207
- Storici F., Lewis LK., Resnick MA. (2001) In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol. 19(8):773-6