Carbocation
A carbocation (/ˌkɑːrboʊˈkætaɪən/[1]) is an ion with a positively charged carbon atom. Among the simplest examples are the methenium CH+
3, methanium CH+
5 and vinyl C
2H+
3 cations. Occasionally, carbocations that bear more than one positively charged carbon atom are also encountered (e.g., ethylene dication C
2H2+
4).[2]


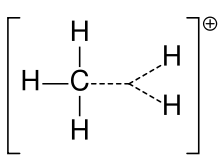
Until the early 1970s, all carbocations were called carbonium ions.[3] In the present-day definition given by the IUPAC, a carbocation is any even-electron cation with significant partial positive charge on a carbon atom. They are further classified in two main categories according to the coordination number of the charged carbon: three in the carbenium ions and five in the carbonium ions. This nomenclature was proposed by G. A. Olah.[4] Carbonium ions, as originally defined by Olah, are characterized by a three-center two-electron delocalized bonding scheme and are essentially synonymous with so-called 'nonclassical carbocations', which are carbocations that contain bridging C–C or C–H σ-bonds. However, others have more narrowly defined the term 'carbonium ion' as formally protonated or alkylated alkanes (i.e., CR5+, where R is hydrogen or alkyl), to the exclusion of nonclassical carbocations like the 2-norbornyl cation.[5]
Definitions
According to the IUPAC, a carbocation is any cation containing an even number of electrons in which a significant portion of the positive charge resides on a carbon atom.[6] Prior to the observation of five-coordinate carbocations by Olah and coworkers, carbocation and carbonium ion were used interchangeably. Olah proposed a redefinition of carbonium ion as a carbocation featuring any type of three-center two-electron bonding, while a carbenium ion was newly coined to refer to a carbocation containing only two-center two-electron bonds with a three-coordinate positive carbon. Subsequently, others have used the term carbonium ion more narrowly to refer to species that are derived (at least formally) from electrophilic attack of H+ or R+ on an alkane, in analogy to other main group onium species, while a carbocation that contains any type of three-centered bonding is referred to as a nonclassical carbocation. In this usage, 2-norbornyl cation is not a carbonium ion, because it is formally derived from protonation of an alkene (norbornene) rather than an alkane, although it is a nonclassical carbocation due to its bridged structure. The IUPAC acknowledges the three divergent definitions of carbonium ion and urges care in the usage of this term. For the remainder of this article, the term carbonium ion will be used in this latter restricted sense, while nonclassical carbocation will be used to refer to any carbocation with C–C and/or C–H σ-bonds delocalized by bridging.
Since the late 1990s, most textbooks have stopped using the term carbonium ion for the classical three-coordinate carbocation. However, some university-level textbooks continue to use the term carbocation as if it were synonymous with carbenium ion,[7][8] or discuss carbocations with only a fleeting reference to the older terminology of carbonium ions[9] or carbenium and carbonium ions.[10] One textbook retains the older name of carbonium ion for carbenium ion to this day, and uses the phrase hypervalent carbonium ion for CH+
5.[11]
A carbocation with a two-coordinate sp-hybridized positive carbon is known as a vinyl cation, while a two-coordinate approximately sp2-hybridized cation resulting from the formal removal of a hydride ion from an arene is termed an aryl cation. These carbocations are very unstable (aryl cations especially so) and are infrequently encountered. Hence, they are frequently omitted from introductory and intermediate level textbooks. The IUPAC definition stipulates that carbocations are even-electron species; hence, radical cations like CH4•+ that are frequently encountered in mass spectrometry are not considered to be carbocations.
History
The history of carbocations dates back to 1891 when G. Merling[12] reported that he added bromine to tropylidene (cycloheptatriene) and then heated the product to obtain a crystalline, water-soluble material, C
7H
7Br. He did not suggest a structure for it; however, Doering and Knox[13] convincingly showed that it was tropylium (cycloheptatrienylium) bromide. This ion is predicted to be aromatic by Hückel's rule.
In 1902, Norris and Kehrman independently discovered that colorless triphenylmethanol gives deep-yellow solutions in concentrated sulfuric acid. Triphenylmethyl chloride similarly formed orange complexes with aluminium and tin chlorides. In 1902, Adolf von Baeyer recognized the salt-like character of the compounds formed. Trityl carbocation (shown below) as a stable carbocationic system has been used as homogeneous organocatalyst in organic synthesis.[14]

He dubbed the relationship between color and salt formation halochromy, of which malachite green is a prime example.
Carbocations are reactive intermediates in many organic reactions. This idea, first proposed by Julius Stieglitz in 1899,[15] was further developed by Hans Meerwein in his 1922 study[16][17] of the Wagner–Meerwein rearrangement. Carbocations were also found to be involved in the SN1 reaction, the E1 reaction, and in rearrangement reactions such as the Whitmore 1,2 shift. The chemical establishment was reluctant to accept the notion of a carbocation and for a long time the Journal of the American Chemical Society refused articles that mentioned them.
The first NMR spectrum of a stable carbocation in solution was published by Doering et al.[18] in 1958. It was the heptamethylbenzenium ion, made by treating hexamethylbenzene with methyl chloride and aluminium chloride. The stable 7-norbornadienyl cation was prepared by Story et al. in 1960[19] by reacting norbornadienyl chloride with silver tetrafluoroborate in sulfur dioxide at −80 °C. The NMR spectrum established that it was non-classically bridged (the first stable non-classical ion observed).
In 1962, Olah directly observed the tert-butyl carbocation by nuclear magnetic resonance as a stable species on dissolving tert-butyl fluoride in magic acid. The NMR of the norbornyl cation was first reported by Schleyer et al.[20] and it was shown to undergo proton-scrambling over a barrier by Saunders et al.[21]
Structure and properties
Carbonium ions can be thought of as protonated alkanes. Although alkanes are usually considered inert, under superacid conditions (e.g., HF/SbF5), the C-H sigma bond can act as a donor to H+. This results in a species that contains a 3c-2e bond between a carbon and two hydrogen atoms, a type of bonding common in boron chemistry, though relatively uncommon for carbon. As an alternative view point, the 3c-2e bond of carbonium ions could be considered as a molecule of H2 coordinated to a carbenium ion. Indeed, carbonium ions frequently decompose by loss of molecular hydrogen to form the corresponding carbenium ion. Structurally, the methanium ion CH5+ is computed to have a minimum energy structure of Cs symmetry. However, the various possible structures of the ion are close in energy and separated by shallow barriers. Hence, the structure of the ion is often described as fluxional. Although there appear to be five bonds to carbon in carbonium ions, they are not hypervalent, as the electron count around the central carbon is only eight, on account of the 3c-2e bond.
The charged carbon atom in a carbenium ion is a "sextet", i.e. it has only six electrons in its outer valence shell instead of the eight valence electrons that ensures maximum stability (octet rule). Therefore, carbocations are often reactive, seeking to fill the octet of valence electrons as well as regain a neutral charge. In accord with VSEPR and Bent's rule, unless geometrically constrained to be pyramidal (e.g., 1-adamantyl cation), 3-coordinate carbenium ions are usually trigonal planar, with a pure p character empty orbital as its lowest unoccupied molecular orbital and CH/CC bonds formed from C(sp2) orbitals. A prototypical example is the methyl cation, CH+
3. For the same reasons, carbocations that are 2-coordinate (vinyl cations) are generally linear in geometry, with CH/CC bonds formed from C(sp) orbitals.
.png)
Alkyl-substituted carbocations follow the order 3° > 2° > 1° > methyl in stability, as can be inferred by the hydride ion affinity values (231, 246, 273, and 312 kcal/mol for (CH3)3C+, (CH3)2CH+, C2H5+, and CH3+).[22] The effect of alkyl substitution is a strong one: tertiary cations are stable and many are directly observable in superacid media, but secondary cations are usually transient and only the isopropyl, s-butyl, and cyclopentyl cations have been observed in solution.[23] There is seldom any experimental support for primary carbocations in the solution phase, even as transient intermediates (the ethyl cation has been proposed for reactions in 99.9% sulfuric acid and in FSO2OH-SbF5),[24] and methyl cation has only been unambiguously identified in the gas phase. In most, if not all cases, the ground state of alleged primary carbocations consist of bridged structures in which positive charge is shared by two or more carbon atoms and are better described as side-protonated alkenes, edge-protonated cyclopropanes, or corner-protonated cyclopropanes rather than true primary cations.[25][26] Even the simple ethyl cation, C2H5+, has been demonstrated experimentally and computationally to be bridged and can be thought of as a symmetrically protonated ethylene molecule. The same is true for higher homologues like n-propyl cation.[27] Neopentyl derivatives are thought to ionize with concomitant migration of a methyl group (anchimeric assistance); thus, in most if not all cases, a discrete neopentyl cation is not believed to be involved.[28]
The stabilization by alkyl groups is explained by hyperconjugation. The donation of electron density from a β C-H or C-C bond into the unoccupied p orbital of the carbocation (a σCH/CC → p interaction) allows the positive charge to be delocalized.
Based on hydride ion affinity, the parent vinyl cation is less stable than even a primary sp2-hybridized carbocation, while an α alkyl-substituted vinyl cation has a stability that is comparable to the latter. Hence, vinyl cations are relatively uncommon intermediates. They can be generated by the ionization of a vinyl electrophile, provided the leaving group is sufficiently good (e.g., TfO–, IPh, or N2). They have been implicated as intermediates in some vinyl substitution reactions (designated as SN1(vinyl)) and as intermediates in the electrophilic addition reactions of arylalkynes. Aryl cations are even more unstable, due to the geometry enforced distortion (spx-character) of the unoccupied orbital. Only N2 in aryldiazonium salts is a good enough leaving group for the chemical generation of aryl cations.
Alkynyl cations are extremely unstable, much less stable than even CH3+ (hydride ion affinity 386 kcal/mol vs. 312 kcal/mol for CH3+) and cannot be generated by purely chemical means. They can, however, be generated radiochemically via the beta decay of tritium: (RC≡CT → [RC≡C3He]+ + e– + νe → RC≡C+ + 3He + e– + νe).[29]

In terms of reactivity, carbocations are susceptible to attack by nucleophiles, like water, alcohols, carboxylates, azide, and halide ions, to form the addition product. Strongly basic nucleophiles, especially hindered ones, favor elimination over addition. Because even weak nucleophiles will react with carbocations, most can only be directly observed or isolated in non-nucleophilic media like superacids.

Carbocations typically undergo rearrangement reactions from less stable structures to equally stable or more stable ones by migration of an alkyl group or hydrogen to the cationic center to form a new carbocationic center. This often occurs with rate constants in excess of 1010 s−1 at ambient temperature and still takes place rapidly (compared to the NMR timescale) at temperatures as low as –120 °C (see Wagner-Meerwein shift). In especially favorable cases like the 2-norbornyl cation, hydrogen shifts may still take place at rates fast enough to interfere with X-ray crystallography at 86 K (–187 °C).[30] Typically, carbocations will rearrange to give a tertiary isomer. For instance, all isomers of C6H12+ rapidly rearrange to give the 1-methyl-1-cyclopentyl cation. This fact often complicates synthetic pathways. For example, when 3-pentanol is heated with aqueous HCl, the initially formed 3-pentyl carbocation rearranges to a statistical mixture of the 3-pentyl and 2-pentyl. These cations react with chloride ion to produce about 1⁄3 3-chloropentane and 2⁄3 2-chloropentane. The Friedel-Crafts alkylation suffers from this limitation; for this reason, the acylation (followed by Wolff-Kishner or Clemmensen reduction to give the alkylated product) is more frequently applied.
A carbocation may be stabilized by resonance by a carbon-carbon double bond next to the ionized carbon. Such cations as allyl cation CH2=CH–CH2+ and benzyl cation C6H5–CH2+ are more stable than most other carbocations due to donation of electron density from π systems to the cationic center. Molecules that can form allyl or benzyl carbocations are especially reactive. These carbocations where the C+ is adjacent to another carbon atom that has a double or triple bond have extra stability because of the overlap of the empty p orbital of the carbocation with the p orbitals of the π bond. This overlap of the orbitals allows the positive charge to be dispersed and electron density from the π system to be shared with the electron-deficient center, resulting in stabilization. The doubly- and triply-benzylic carbocations, diphenylcarbenium and triphenylcarbenium (trityl) cation, are particularly stable. For the same reasons, the partial p character of strained C–C bonds in cyclopropyl groups also allows for donation of electron density and stabilizes the cyclopropylmethyl (cyclopropylcarbinyl) cation.
As noted in the history section, the tropylium cation (C7H7+) was one of the first carbocations to be discovered, due to its aromatic stability. On the other hand, the anti-aromatic cyclopentadienylium cation (C5H5+) is destabilized by some 50 kcal/mol. The cyclopropenium cation (C3H3+), although somewhat destabilized by angle strain, is still clearly stabilized by aromaticity when compared to its open-chain analog, allyl cation.
The stability order of carbocations, from most stable to least stable as reflected by hydride ion affinity (HIA) values, are as follows (HIA values in kcal/mol in parentheses):
| Carbocation | c-C7H7+ (most stable) | (C6H5)3C+ | c-C3H3+ | (C6H5)2CH+ | 2-norbornyl+ | t-C4H9+ | C6H5CH2+ | iso-C3H7+ |
| HIA (kcal/mol) | 201 | 215 | 221 | 222 | 231 | 231 | 234 | 246 |
| Carbocation | (c-C3H5)CH2+ | CH2=CH–CH2+ | c-C5H5+ | CH≡C–CH2+ | C2H5+ | CH2=CH+ | C6H5+ | CH3+ (least stable) |
| HIA (kcal/mol) | 249 | 256 | 258 | 270 | 273 | 287 | 298 | 312 |
The effect of hyperconjugation is strongly stabilizing for carbocations: hyperconjugation with alkyl substituents is often as stabilizing or even more so than conjugation with a π system. Although conjugation to unsaturated groups results in significant stabilization by the mesomeric effect (resonance), the benefit is partially offset by the presence of a more electronegative sp2 or sp carbon next to the carbocationic center. Thus, as reflected by hydride ion affinities, a secondary carbocation is more stabilized than the allyl cation, while a tertiary carbocation is more stabilized than the benzyl cation, results that may seem counterintuitive on first glance.
Oxocarbenium and iminium ions have important secondary canonical forms (resonance structures) in which carbon bears a positive charge. As such, they are carbocations according to the IUPAC definition although some chemists do not regard them to be "true" carbocations, as their most important resonance contributors carry the formal positive charge on an oxygen or nitrogen atom, respectively.
Non-classical ions
Some carbocations such as the 2-norbornyl cation exhibit more or less symmetrical three-center two-electron bonding. Such structures, referred to as non-classical carbocations, involve the delocalization of the bonds involved in the σ-framework of the molecule and the drawing of "no-bond" resonance forms (beyond the relatively simple variety encountered in hyperconjugation). As circumstantial evidence of its unusual bonding, the 2-norbornyl cation is also more stable than a typical "secondary" carbocation, being roughly as stable as t-butyl cation, according to hydride ion affinity.
The existence of non-classical carbocations was once the subject of great controversy. On opposing sides were Brown, who believed that the what appeared to be a non-classical carbocation represents the average of two rapidly equilibrating classical species (or possibly two structures exhibiting some degree of bridging or leaning but is nevertheless not symmetric) and that the true non-classical structure is a transition state between the two potential energy minima, and Winstein, who believed that a non-classical structure that possessed a plane of symmetry was the sole potential energy minimum and that the classical structures merely two contributing resonance forms of this non-classical species. George Olah's discovery of superacidic media to allow carbocations to be directly observed, together with a very sensitive NMR technique developed by Martin Saunders to distinguish between the two scenarios, played important roles in resolving this controversy.[31][32] At least for the 2-norbornyl cation itself, the controversy has been settled overwhelmingly in Winstein's favor, with no sign of the putative interconverting classical species, even at temperatures as low as 6 K, and a 2013 crystal structure showing a distinctly non-classical structure.[33][30] A variety of carbocations (e.g., ethyl cation, see above) are now believed to adopt non-classical structures. However, in many cases, the energy difference between the two possible "classical" structures and the "non-classical" one is very small, and it may be difficult to distinguish between the two possibilities experimentally.
Specific carbocations
Cyclopropylcarbinyl cations can be studied by NMR:[34][35]
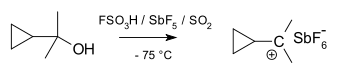
In the NMR spectrum of a dimethyl derivative, two nonequivalent signals are found for the two methyl groups, indicating that the molecular conformation of this cation is not perpendicular (as in A), which possesses a mirror plane, but is bisected (as in B) with the empty p-orbital parallel to the cyclopropyl ring system:
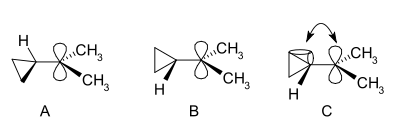
In terms of bent bond theory, this preference is explained by assuming favorable orbital overlap between the filled cyclopropane bent bonds and the empty p-orbital.[36]
Pyramidal carbocation
| Pyramidal Carbocations | ||
|---|---|---|
 |
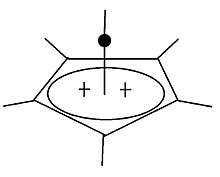 |
Besides the classical and non-classical carbocations, a third class can be distinguished: pyramidal carbocations. In these ions, a single carbon atom hovers over a four- or five-sided polygon, in effect forming a pyramid. The square pyramidal ion will carry a charge of +1, the Pentagonal pyramidal ion will carry +2.
A stable hexagonal-pyramidal configuration of tropylium trication, (C7H7)3+, has been also predicted.[37] In this case, the coordination number of carbon reaches seven. The crystal structure of [C6(CH3)6][SbF6]2•HSO3F confirms the pentagonal-pyramidal shape of the hexamethylbenzene dication.[38] |
| An example of the monovalent carbocation | An example of the divalent carbocation |
See also
References
- "Carbocation". Oxford Dictionaries UK Dictionary. Oxford University Press. Retrieved 2016-01-21.
- Grützmacher, Hansjörg; Marchand, Christina M. (1997). "Heteroatom stabilized carbenium ions". Coord. Chem. Rev. 163: 287–344. doi:10.1016/S0010-8545(97)00043-X.
- Robert B. Grossman (2007-07-31). The Art of Writing Reasonable Organic Reaction Mechanisms. Springer Science & Business Media. pp. 105–. ISBN 978-0-387-95468-4.
- Olah, George A. (1972). "Stable carbocations. CXVIII. General concept and structure of carbocations based on differentiation of trivalent (classical) carbenium ions from three-center bound penta- of tetracoordinated (nonclassical) carbonium ions. Role of carbocations in electrophilic reactions". J. Am. Chem. Soc. 94 (3): 808–820. doi:10.1021/ja00758a020.
- Sommer, J.; Jost, R. (2000-01-01). "Carbenium and carbonium ions in liquid- and solid-superacid-catalyzed activation of small alkanes". Pure and Applied Chemistry. 72 (12): 2309–2318. doi:10.1351/pac200072122309. ISSN 1365-3075.
- Chemistry, International Union of Pure and Applied (2009), "Carbocation", IUPAC Compendium of Chemical Terminology, IUPAC, doi:10.1351/goldbook.C00817, ISBN 978-0967855097, retrieved 2018-11-03
- McMurry, John. Organic chemistry (5th ed.). ISBN 978-0-534-37617-8.
- Vollhardt, K. Peter C.; Schore, Neil Eric (2018). Organic chemistry: Structure and function (8th ed.). New York. ISBN 9781319079451. OCLC 1007924903.
- Yurkanis Bruice, Paula (2004). Organic Chemistry (4th ed.). ISBN 978-0-13-140748-0.
- Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2001). Organic Chemistry (1st ed.). Oxford University Press. ISBN 978-0-19-850346-0.
- Fox, Marye Anne; Whitesell, James K. (1997). Organic Chemistry. ISBN 978-0-7637-0413-1.
- Merling, G. (1891). "Ueber Tropin". Berichte der Deutschen Chemischen Gesellschaft. 24 (2): 3108–3126. doi:10.1002/cber.189102402151. ISSN 0365-9496.
- Doering, W. von E.; Knox, L. H. (1954). "The Cycloheptatrienylium (Tropylium) Ion". J. Am. Chem. Soc. 76 (12): 3203–3206. doi:10.1021/ja01641a027.
- "Discovery of an in situ carbocationic system using trityl chloride as a homogeneous organocatalyst". Tetrahedron. 69: 212–218. 2013. doi:10.1016/j.tet.2012.10.042.
- "On the Constitution of the Salts of Imido-Ethers and other Carbimide Derivatives". Am. Chem. J. 21: 101. ISSN 0096-4085.
- Meerwein, H.; Emster, K. van (1922). "About the equilibrium isomerism between bornyl chloride isobornyl chloride and camphene chlorohydrate". Berichte. 55: 2500.
- Rzepa, H. S.; Allan, C. S. M. (2010). "Racemization of Isobornyl Chloride via Carbocations: A Nonclassical Look at a Classic Mechanism". Journal of Chemical Education. 87 (2): 221. Bibcode:2010JChEd..87..221R. doi:10.1021/ed800058c.
- Doering, W. von E.; Saunders, M.; Boyton, H. G.; Earhart, H. W.; Wadley, E. F.; Edwards, W. R.; Laber, G. (1958). "The 1,1,2,3,4,5,6-heptamethylbenzenonium ion". Tetrahedron. 4 (1–2): 178–185. doi:10.1016/0040-4020(58)88016-3.
- Story, Paul R.; Saunders, Martin (1960). "The 7-norbornadienyl carbonium ion". J. Am. Chem. Soc. 82 (23): 6199. doi:10.1021/ja01508a058.
- Schleyer, Paul von R.; Watts, William E.; Fort, Raymond C.; Comisarow, Melvin B.; Olah, George A. (1964). "Stable Carbonium Ions. X.1 Direct Nuclear Magnetic Resonance Observation of the 2-Norbornyl Cation". J. Am. Chem. Soc. 86 (24): 5679–5680. doi:10.1021/ja01078a056.
- Saunders, Martin; Schleyer, Paul von R.; Olah, George A. (1964). "Stable Carbonium Ions. XI.1 The Rate of Hydride Shifts in the 2-Norbornyl Cation". J. Am. Chem. Soc. 86 (24): 5680–5681. doi:10.1021/ja01078a057.
- Anslyn, Eric V.; Dougherty, Dennis A. (2000). Modern Physical Organic Chemistry. Sausalito, CA: University Science Books. ISBN 978-1891389313.
- A., Carroll, Felix (2010). Perspectives on structure and mechanism in organic chemistry (2nd ed.). Hoboken, N.J.: John Wiley. ISBN 9780470276105. OCLC 286483846.
- Olah, George A.; O'Brien, Daniel H.; White, Anthony Mallinson. (October 1967). "Stable carbonium ions. LII. Protonated esters and their cleavage in fluorosulfonic acid-antimony pentafluoride solution". Journal of the American Chemical Society. 89 (22): 5694–5700. doi:10.1021/ja00998a036. ISSN 0002-7863.
- 1937-, Carey, Francis A. (2007). Advanced organic chemistry. Sundberg, Richard J., 1938- (5th ed.). New York: Springer. ISBN 9780387448978. OCLC 154040953.CS1 maint: numeric names: authors list (link)
- H., Lowry, Thomas (1987). Mechanism and theory in organic chemistry. Richardson, Kathleen Schueller. (3rd ed.). New York: Harper & Row. ISBN 0060440848. OCLC 14214254.
- Schultz, Jocelyn C.; Houle, F. A.; Beauchamp, J. L. (July 1984). "Photoelectron spectroscopy of 1-propyl, 1-butyl, isobutyl, neopentyl, and 2-butyl radicals: free radical precursors to high-energy carbonium ion isomers". Journal of the American Chemical Society. 106 (14): 3917–3927. doi:10.1021/ja00326a006. ISSN 0002-7863.
- Yamataka, Hiroshi; Ando, Takashi; Nagase, Shigeru; Hanamura, Mitsuyasu; Morokuma, Keiji (February 1984). "Ab initio MO calculations of isotope effects in model processes of neopentyl ester solvolysis". The Journal of Organic Chemistry. 49 (4): 631–635. doi:10.1021/jo00178a010. ISSN 0022-3263.
- Angelini, Giancarlo.; Hanack, Michael.; Vermehren, Jan.; Speranza, Maurizio. (1988-02-17). "Generation and trapping of an alkynyl cation". Journal of the American Chemical Society. 110 (4): 1298–1299. doi:10.1021/ja00212a052. ISSN 0002-7863.
- Scholz, F.; Himmel, D.; Heinemann, F. W.; Schleyer, P. v R.; Meyer, K.; Krossing, I. (2013-07-05). "Crystal Structure Determination of the Nonclassical 2-Norbornyl Cation". Science. 341 (6141): 62–64. Bibcode:2013Sci...341...62S. doi:10.1126/science.1238849. ISSN 0036-8075. PMID 23828938.
- Olah, George A.; Prakash, G. K. Surya; Saunders, Martin (May 2002). "Conclusion of the classical-nonclassical ion controversy based on the structural study of the 2-norbornyl cation". Accounts of Chemical Research. 16 (12): 440–448. doi:10.1021/ar00096a003.
- George A. Olah - Nobel Lecture
- Yannoni, C. S.; Myhre, P. C.; Webb, Gretchen G. (November 1990). "Magic angle spinning nuclear magnetic resonance near liquid-helium temperatures. Variable-temperature CPMAS spectra of the 2-norbornyl cation to 6 K". Journal of the American Chemical Society. 112 (24): 8991–8992. doi:10.1021/ja00180a060. ISSN 0002-7863.
- Kabakoff, David S.; Namanworth, Eli (1970). "Nuclear magnetic double resonance studies of the dimethylcyclopropylcarbinyl cation. Measurement of the rotation barrier". J. Am. Chem. Soc. 92 (10): 3234–3235. doi:10.1021/ja00713a080.
- Pittman Jr., Charles U.; Olah, George A. (1965). "Stable Carbonium Ions. XVII.1a Cyclopropyl Carbonium Ions and Protonated Cyclopropyl Ketones". J. Am. Chem. Soc. 87 (22): 5123–5132. doi:10.1021/ja00950a026.
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry Part A (2nd ed.).
- Wang, George; Rahman, A. K. Fazlur; Wang, Bin (May 2018). "Ab initio calculations of ionic hydrocarbon compounds with heptacoordinate carbon". Journal of Molecular Modeling. 24 (5): 116. doi:10.1007/s00894-018-3640-9. ISSN 1610-2940. PMID 29696384.
- Malischewski, Moritz; Seppelt, K. (2016-11-25). "Crystal Structure Determination of the Pentagonal-Pyramidal Hexamethylbenzene Dication C6(CH3)62+". Angewandte Chemie International Edition. 56 (1): 368–370. doi:10.1002/anie.201608795. ISSN 1433-7851. PMID 27885766.
External links

- Press Release The 1994 Nobel Prize in Chemistry". Nobelprize.org. 9 Jun 2010