Coordination number
In chemistry, crystallography, and materials science the coordination number, also called ligancy, of a central atom in a molecule or crystal is the number of atoms, molecules or ions bonded to it. The ion/molecule/atom surrounding the central ion/molecule/atom is called a ligand. This number is determined somewhat differently for molecules than for crystals.
For molecules and polyatomic ions the coordination number of an atom is determined by simply counting the other atoms to which it is bonded (by either single or multiple bonds).[1] For example, [Cr(NH3)2Cl2Br2]− has Cr3+ as its central cation, and has a coordination number of 6.
However the solid-state structures of crystals often have less clearly defined bonds, and in these cases a count of neighboring atoms is employed. The simplest method is one used in materials science. The usual value of the coordination number for a given structure refers to an atom in the interior of a crystal lattice with neighbors in all directions. In contexts where crystal surfaces are important, such as materials science and heterogeneous catalysis, the value for an interior atom is the bulk coordination number, while the value for an atom at a surface of the crystal is the surface coordination number.
Molecules, polyatomic ions and coordination complexes
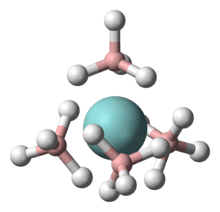
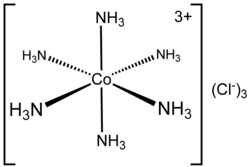
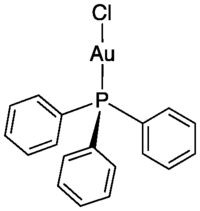
In chemistry, coordination number (C.N.), defined originally in 1893 by Alfred Werner, is the total number of neighbors of a central atom in a molecule or ion.[1][3] Although a carbon atom has four chemical bonds in most stable molecules, the coordination number of each carbon is four in methane (CH4), three in ethylene (H2C=CH2, each C is bonded to 2H + 1C = 3 atoms), and two in acetylene (HC≡CH). In effect we count the first bond (or sigma bond) to each neighboring atom, but not the other bonds (pi bonds).
In coordination complexes, only the first or sigma bond between each ligand and the central atom counts, but not any pi bonds to the same ligands. In tungsten hexacarbonyl, W(CO)6, the coordination number of tungsten (W) is counted as six although pi as well as sigma bonding is important in such metal carbonyls.
The most common coordination number for d-block transition metal complexes is 6, with an octahedral geometry. The observed range is 2 (e.g., AuI in Ph3PAuCl) to 9 (e.g., ReVII in [ReH9]2−). Metals in the f-block (the lanthanoids and actinoids) can accommodate higher coordination number due to their greater ionic radii and availability of more orbitals for bonding. Coordination numbers of 8 to 12 are commonly observed for f-block elements. For example, with bidentate nitrate ions as ligands, CeIV and ThIV form the 12-coordinate ions [Ce(NO3)6]2− (ceric ammonium nitrate) and [Th(NO3)6]2−. When the surrounding ligands are much smaller than the central atom, even higher coordination numbers may be possible. One computational chemistry study predicted a particularly stable PbHe2+
15 ion composed of a central lead ion coordinated with no fewer than 15 helium atoms.[4] At the opposite extreme, steric shielding can give rise to unusually low coordination numbers. An extremely rare instance of a metal adopting a coordination number of 1 occurs in the terphenyl-based arylthallium(I) complex 2,6-Tipp2C6H3Tl, where Tipp is the 2,4,6-triisopropylphenyl group.[5]
For π-electron ligands such as the cyclopentadienide ion [C5H5]−, alkenes and the cyclooctatetraenide ion [C8H8]2−, the number of atoms in the π-electron system that bind to the central atom is termed the hapticity.[6] In ferrocene the hapticity, η, of each cyclopentadienide anion is five, Fe(η5-C5H5)2. There are various ways of assigning the contribution made to the coordination number of the central iron atom by each cyclopentadienide ligand. The contribution could be assigned as one since there is one ligand, or as five since there are five neighbouring atoms, or as three since there are three electron pairs involved. Normally the count of electron pairs is taken.[7]
Crystalline substances
In order to determine the coordination number of an atom in a crystal, the crystal structure has first to be determined. This is achieved using X-ray, neutron or electron diffraction.[8] Once the positions of the atoms within the unit cell of the crystal are known the coordination number of an atom can be determined. For molecular solids or coordination complexes the units of the polyatomic species can be detected and a count of the bonds can be performed. Solids with lattice structures which includes metals and many inorganic solids can have regular structures where coordinating atoms are all at the same distance and they form the vertices of a coordination polyhedron. However, there are also many such solids where the structures are irregular. In materials science, the bulk coordination number of a given atom in the interior of a crystal lattice is the number of nearest neighbours to a given atom. For an atom at a surface of a crystal, the surface coordination number is always less than the bulk coordination number. The surface coordination number is dependent on the Miller indices of the surface. In a body-centered cubic (BCC) crystal, the bulk coordination number is 8, whereas, for the (100) surface, the surface coordination number is 4.
α-Aluminium has a regular cubic close packed structure, fcc, where each aluminium atom has 12 nearest neighbors, 6 in the same plane and 3 above and below and the coordination polyhedron is a cuboctahedron. α-Iron has a body centered cubic structure where each iron atom has 8 nearest neighbors situated at the corners of a cube.

The two most common allotropes of carbon have different coordination numbers. In diamond, each carbon atom is at the centre of a regular tetrahedron formed by four other carbon atoms, the coordination number is four, as for methane. Graphite is made of two-dimensional layers in which each carbon is covalently bonded to three other carbons; atoms in other layers are further away and are not nearest neighbours, giving a coordination number of 3.[9]
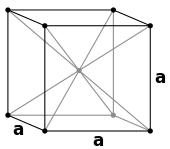
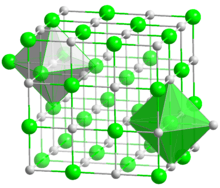
For chemical compounds with regular lattices such as sodium chloride and caesium chloride, a count of the nearest neighbors gives a good picture of the environment of the ions. In sodium chloride each sodium ion has 6 chloride ions as nearest neighbours (at 276 pm) at the corners of an octahedron and each chloride ion has 6 sodium atoms (also at 276 pm) at the corners of an octahedron. In caesium chloride each caesium has 8 chloride ions (at 356 pm) situated at the corners of a cube and each chloride has eight caesium ions (also at 356 pm) at the corners of a cube.
However, not all crystal structures are regular and the neighboring atoms may not all be at the same distance. In these cases a different definition of coordination number is used that includes atoms at a greater distance than the nearest neighbours. The very broad definition adopted by the International Union of Crystallography, IUCR, states that the coordination number of an atom in a crystalline solid depends on the chemical bonding model and the way in which the coordination number is calculated.[10][11]
Some metals have irregular structures. For example, zinc has a distorted hexagonal close packed structure. Regular hexagonal close packing of spheres would predict that each atom has 12 nearest neighbours and a triangular orthobicupola (also called an anticuboctahedron or twinned cuboctahedron) coordination polyhedron.[9][12] In zinc there are only 6 nearest neighbours at 266 pm in the same close packed plane with six other, next-nearest neighbours, equidistant, three in each of the close packed planes above and below at 291 pm. It is considered to be reasonable to describe the coordination number as 12 rather than 6.[11] Similar considerations can be applied to the regular body centred cube structure where in addition to the 8 nearest neighbors there 6 more, approximately 15% more distant,[9] and in this case the coordination number is often considered to be 14.
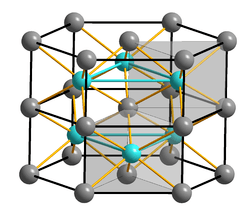
Many chemical compounds have distorted structures. Nickel arsenide, NiAs has a structure where nickel and arsenic atoms are 6-coordinate. Unlike sodium chloride where the chloride ions are cubic close packed, the arsenic anions are hexagonal close packed. The nickel ions are 6-coordinate with a distorted octahedral coordination polyhedron where columns of octahedra share opposite faces. The arsenic ions are not octahedrally coordinated but have a trigonal prismatic coordination polyhedron. A consequence of this arrangement is that the nickel atoms are rather close to each other. Other compounds that share this structure, or a closely related one are some transition metal sulfides such as FeS and CoS, as well as some intermetallics. In cobalt(II) telluride, CoTe, the six tellurium and two cobalt atoms are all equidistant from the central Co atom.[9]
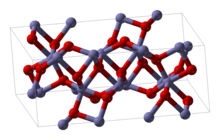
Two other examples of commonly-encountered chemicals are Fe2O3 and TiO2. Fe2O3 has a crystal structure that can be described as having a near close packed array of oxygen atoms with iron atoms filling two thirds of the octahedral holes. However each iron atom has 3 nearest neighbors and 3 others a little further away. The structure is quite complex, the oxygen atoms are coordinated to four iron atoms and the iron atoms in turn share vertices, edges and faces of the distorted octahedra.[9] TiO2 has the rutile structure. The titanium atoms 6-coordinate, 2 atoms at 198.3 pm and 4 at 194.6 pm, in a slightly distorted octahedron. The octahedra around the titanium atoms share edges and vertices to form a 3-D network. The oxide ions are 3-coordinate in a trigonal planar configuration.[13]
Usage in quasicrystal, liquid and other disordered systems
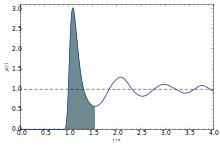
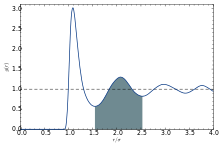
The coordination number of systems with disorder cannot be precisely defined.
The first coordination number can be defined using the radial distribution function g(r):[14][15]

where r0 is the rightmost position starting from r = 0 whereon g(r) is approximately zero, r1 is the first minimum. Therefore, it is the area under the first peak of g(r).
The second coordination number is defined similarly:

Alternative definitions for the coordination number can be found in literature, but in essence the main idea is the same. One of those definition are as follows: Denoting the position of the first peak as rp,

The first coordination shell is the spherical shell with radius between r0 and r1 around the central particle under investigation.
References
- 1 2 IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "coordination number".
- ↑ Haaland, Arne; Shorokhov, Dmitry J.; Tutukin, Andrey V.; Volden, Hans Vidar; Swang, Ole; McGrady, G. Sean; Kaltsoyannis, Nikolas; Downs, Anthony J.; Tang, Christina Y.; Turner, John F. C. (2002). "Molecular Structures of Two Metal Tetrakis(tetrahydroborates), Zr(BH4)4 and U(BH4)4: Equilibrium Conformations and Barriers to Internal Rotation of the Triply Bridging BH4 Groups". Inorganic Chemistry. 41: 6646–6655. doi:10.1021/ic020357z.
- ↑ De, A.K. (2003). A Text Book of Inorganic Chemistry. New Age International Publishers. p. 88. ISBN 8122413846.
- ↑ Hermann, Andreas; Lein, Matthias; Schwerdtfeger, Peter (2007). "The Search for the Species with the Highest Coordination Number". Angewandte Chemie International Edition. 46 (14): 2444. doi:10.1002/anie.200604148. PMID 17315141.
- ↑ Niemeyer, Mark; Power, Philip P. (1998-05-18). "Synthesis and Solid-State Structure of 2,6-Trip2C6H3Tl (Trip=2,4,6-iPr3C6H2): A Monomeric Arylthallium(I) Compound with a Singly Coordinated Thallium Atom". Angewandte Chemie International Edition. 37 (9): 1277–1279. doi:10.1002/(SICI)1521-3773(19980518)37:93.0.CO;2-1. ISSN 1521-3773.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "hapticity".
- ↑ Crabtree, Robert H. (2009). The Organometallic Chemistry of the Transition Metals. John Wiley & Sons. ISBN 9780470257623.
- ↑ Massa, Werner (1999). Crystal Structure Determination (English ed.). Springer. pp. 67–92.
- 1 2 3 4 5 Wells, A.F. (1984). Structural Inorganic Chemistry (5th ed.). Oxford Science Publications. ISBN 0198553706.
- ↑
- 1 2 Mittemeijer, Eric J. (2010). Fundamentals of Materials Science: The Microstructure–Property Relationship using metals as model systems. Springer. ISBN 9783642105005.
- ↑ Piróth, A.; Sólyom, Jenö (2007). Fundamentals of the Physics of Solids: Volume 1: Structure and Dynamics. Springer. p. 227. ISBN 9783540726005.
- ↑ Diebold, Ulrike (2003). "The surface science of titanium dioxide". Surface Science Reports. 48 (5–8): 53–229. Bibcode:2003SurSR..48...53D. doi:10.1016/S0167-5729(02)00100-0. ISSN 0167-5729.
- ↑ Yoshio Waseda, The Structure of Non-crystalline Materials – Liquids and Amorphous Solids, McGraw-Hill, New York, 1980, pp. 48–51.
- ↑ Vahvaselkä, K. S.; Mangs, J. M. (1988). "X-ray diffraction study of liquid sulfur". Physica Scripta. 38 (5): 737. Bibcode:1988PhyS...38..737V. doi:10.1088/0031-8949/38/5/017.